In 1980, posttraumatic stress disorder (PTSD) was first established as a diagnosis in DSM-III under the anxiety disorders category. Although there are important differences between PTSD and the other anxiety disorders—and the proper classification of these disorders continues to be debated—PTSD and other anxiety disorder patients have in common increased levels of anxiety symptoms that are responsive to treatment with benzodiazepine medications. The efficacy of benzodiazepines in the treatment of anxiety disorders has led to an effort to develop animal models, such as inescapable stress, to understand the mechanisms involved.
Animals exposed to chronic inescapable stress develop behaviors that indicate excessive fear and anxiety, such as increased fearfulness, increased defecation, and avoidance of novel situations (e.g., an open field). Exposure to inescapable stressors such as foot shock or forced swim also resulted in a 20%–30% decrease in benzodiazepine receptor binding in the frontal cortex
(1,
2) and cerebral cortex
(3–
7), with some studies showing a decrease in the hippocampus
(3,
4,
6,
7, but see references
2,
5,
8, and
9). Exposure to stress has no effects on benzodiazepine receptor binding in the pons, striatum, thalamus, cerebellum, midbrain, or occipital cortex
(1,
3–
5,
7–
12). One study that used a different model of stressor, defeat stress, showed an increase in benzodiazepine receptor binding in the cortex
(12). These studies are consistent with a role for alterations in benzodiazepine binding in anxiety
(13), with findings showing a specific decrease in the frontal cortex and, although not as consistently, a decrease in the hippocampus. The findings suggest that decreased benzodiazepine receptor binding in the frontal cortex may play a role in the symptoms of PTSD.
Studies in human subjects are also consistent with a relationship between stress and alterations in benzodiazepine receptor binding
(14–
16). FG7142, a benzodiazepine receptor inverse agonist, induced severe anxiety that resembled panic attacks and biological characteristics of anxiety in healthy subjects
(14). Patients with combat-related PTSD did not exhibit a greater behavioral response (i.e., anxiety) with the benzodiazepine receptor antagonist flumazenil than did healthy subjects
(16); however, flumazenil has no pharmacological effect on the receptor per se. Future studies are required with other agents, such as inverse agonists, that have an effect on the benzodiazepine receptor. Benzodiazepine administration has been shown to be efficacious in the treatment of symptoms of chronic PTSD and anxiety
(17–
19), although it has not been shown to prevent the development of chronic PTSD
(20). These findings suggest that alterations in benzodiazepine receptor function may underlie the symptoms of anxiety and, more specifically, PTSD.
Recently, neuroimaging techniques have been developed that provide quantitation of neuroreceptors in living human brain. Neuroreceptor imaging, however, has not yet been applied to the study of PTSD. One area of research that holds promise for the study of anxiety disorders is quantitation of benzodiazepine receptor binding. Iomazenil is a compound that binds with high affinity to the benzodiazepine receptor. Iomazenil
(21–
23) and other compounds
(24) have been used to image the benzodiazepine receptor in patients with other anxiety disorders besides PTSD. Single photon emission computed tomography (SPECT) with [
123I]iomazenil was demonstrated to provide reliable and valid quantitation of benzodiazepine receptors in human subjects
(25–
27). No studies to date have used nuclear imaging to examine central benzodiazepine receptor binding in PTSD. The purpose of this study was to measure benzodiazepine receptor binding by means of SPECT and [
123I]iomazenil in patients with PTSD and matched normal comparison subjects. On the basis of the aforementioned preclinical studies, we hypothesized a decrease in benzodiazepine receptor binding in the frontal cortex and hippocampus, but not in other areas (pons, striatum, thalamus, cerebellum, or midbrain) in patients with PTSD relative to normal comparison subjects.
Method
Thirteen subjects seeking outpatient treatment for Vietnam combat-related PTSD were recruited for the study. All PTSD patients were male, right-handed, and met criteria for PTSD as determined by the Structured Clinical Interview for DSM-IV (SCID)
(28). Eleven (85%) of the 13 patients were white, and two (15%) were black. At study entry, PTSD patients had not been taking any psychotropic medications for at least 4 weeks. The patients had no history of benzodiazepine usage in the preceding 6 months. Urinary toxicology screens were performed, and patients in whom the presence of benzodiazepines or other substances were detected were excluded from the study. PTSD patients with a history of alcohol or drug abuse or dependence in the 6 months preceding the study, meningitis, traumatic brain injury, loss of consciousness of greater than 1 minute, other neurological disorder, or psychotic disorder determined during administration of the SCID were excluded. Patients who had a history of foreign bodies that would preclude MRI scanning were also excluded.
Comparison subjects were healthy subjects without a history of medical or psychiatric disorder who were case-matched to the PTSD patients. Comparison subjects were recruited by newspaper advertisement and were selected to be matched to the patients by race and sex. In addition comparison subjects were selected to be similar in age to PTSD patients. All comparison subjects were male, right-handed, and were not taking any psychotropic medication nor had they in the preceding 4 weeks; 11 (85%) of the 13 comparison subjects were white, and two (15%) were black. There was no significant difference in age between the PTSD patients (mean=49 years, SD=2) and the comparison subjects (mean=44 years, SD=9). In addition to the exclusion criteria outlined above for the PTSD patients, comparison subjects with a history of psychiatric disorder as determined by the non-patient version of the SCID were excluded. All subjects in this study provided written informed consent for participation.
PTSD patients were evaluated with the SCID to determine the presence of psychiatric diagnoses. None of the 13 patients had a history of a psychiatric disorder that preceded the onset of PTSD, and in all cases PTSD was the primary diagnosis. Comorbid conditions detected in the 13 PTSD patients included panic disorder with agoraphobia (current: 54% [N=7]; lifetime: 69% [N=9]), social phobia (current: 31% [N=4]; lifetime: (38% [N=5]), simple phobia (current: 8% [N=1]; lifetime: 15% [N=2]), generalized anxiety disorder (current: 8% [N=1]; lifetime: 15% [N=2]), bulimia (current: 8% [N=1]; lifetime: 15% [N=2]), and major depression (current: 38% [N=5]); lifetime: 85% [N=11]). None of the patients met criteria for current or lifetime anorexia, obsessive-compulsive disorder, schizophrenia, bipolar disorder, somatization disorder, somatic pain disorder, undifferentiated somatization disorder, or hypochondriasis. Two patients (15%) met criteria for past alcohol dependence, and one (8%) met criteria for past alcohol abuse.
All subjects were evaluated at the time of the scan with the Panic Attack Symptom Scale
(29), Hamilton Anxiety Rating Scale, and the Brief Psychiatric Rating Scale (BPRS). PTSD patients had higher levels of anxiety symptoms than the comparison subjects, as measured by the Panic Attack Symptom Scale (mean=3.7 [SD=4.4] versus 1.4 [SD=2.9], respectively) and the Hamilton anxiety scale (mean=17.3 [SD=8.2] versus 2.9 [SD=4.2]); they also had higher levels of general psychiatric symptoms as measured with the BPRS (mean=29.3 [SD=5.8] versus 19.4 [SD=1.1]).
All subjects underwent a single SPECT [
123I]iomazenil scan for measurement of benzodiazepine receptors with the equilibrium technique on a Ceraspect camera (Digital Scintigraphics, Inc., Waltham, Mass.). The methods of the equilibrium technique and properties of the Ceraspect camera have been previously described in detail
(25–
27). Briefly, a bolus dose of 6 mCi [
123I]iomazenil was administered followed by a continuous infusion of radiotracer over 8 hours (ratio of bolus/infusion rate=3.8). Starting at 6 hours after administration of the bolus, subjects underwent SPECT scanning once radioligand uptake into the brain reached equilibrium, as determined by no change in brain tissue concentration over three consecutive scans. At equilibrium (as determined by this method), rate of transfer of radioligand (i.e., [
123I]iomazenil) onto the receptor is assumed to be equal to rate of transfer off of receptor. Subjects underwent three 10-minute acquisitions. SPECT images were reconstructed by using methods previously described
(25–
27). Venous blood was drawn at the time of the image acquisition (i.e., 6 hours after initiation of bolus plus constant infusion protocol) for measurement of total and free (or non-protein-bound) [
123I]iomazenil parent compound in plasma by using methods previously described
(30). There was no difference between PTSD and comparison subjects in plasma concentrations of free non-protein-bound parent compound (mean=0.0014 [SD=0.0008] and 0.0012 [SD=0.0002]μCi/ml, respectively), total parent compound (mean=0.0041 [SD=0.0024] and 0.0035 [SD=0.0009] μCi/ml), or in the fraction of free parent in plasma (mean=0.34 [SD=0.06] and 0.32 [SD=0.02]).
Magnetic resonance (MR) images were obtained in all subjects for coregistration with SPECT. The MR acquisition protocol, method for coregistration, and use of MR for identification of regions of interest on functional images have been previously described in detail
(31).
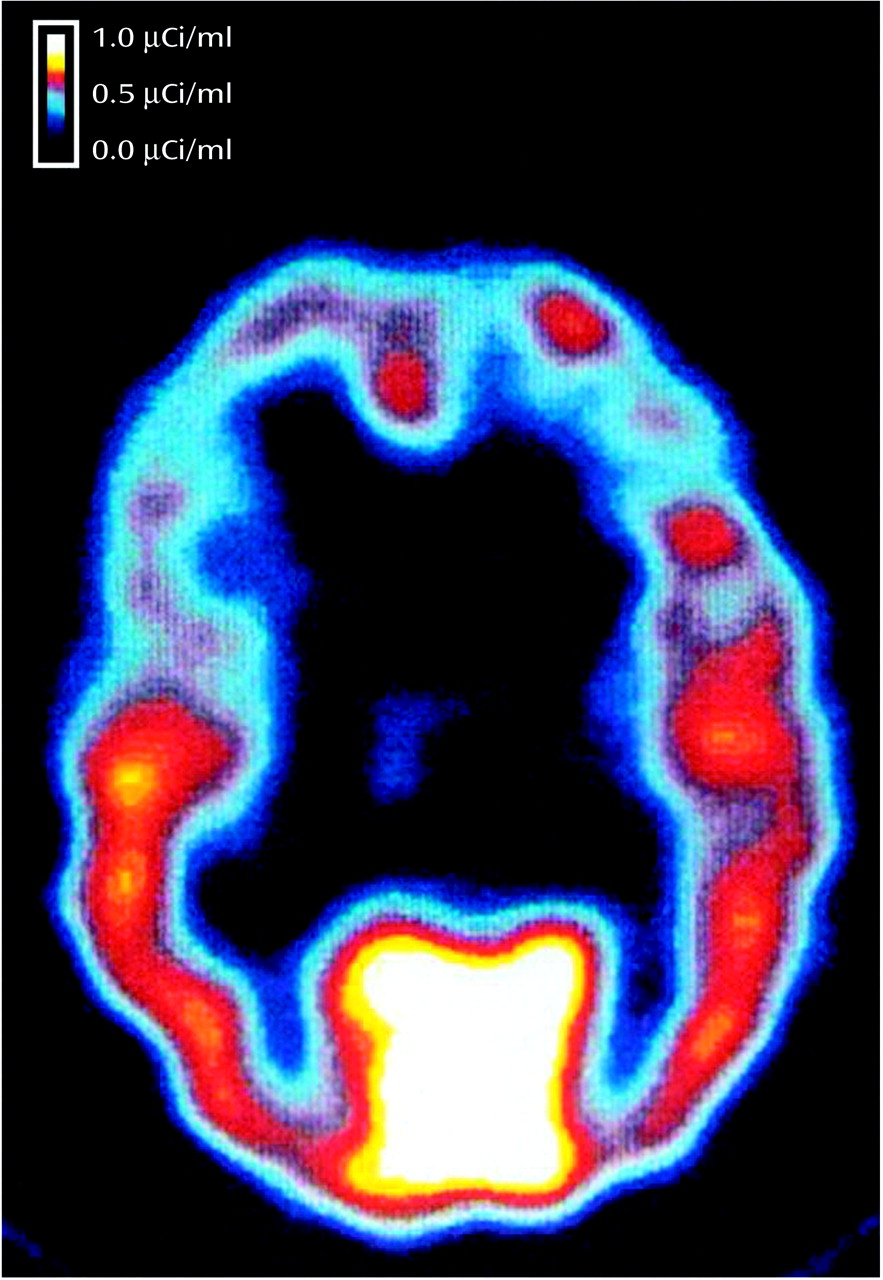
Global radioactivity in the three scans was examined to determine that the patient was at equilibrium (retrospectively determined by the fact that there was not greater than 10% difference in global distribution volume between the three scans). This criterion was used to determine equilibrium for all subjects (consistent with our prior findings of the time course to equilibrium). The three SPECT scans were realigned to the first scan in the series and summed into a single image (
Figure 1). SPECT scans were then converted from counts per pixel per minute to μCi/ml by using calibration factors based on phantom studies. Distribution volumes, which are equal to the ratio of tissue concentration of radiotracer to plasma concentration of non-protein-bound parent radiotracer at equilibrium, were then calculated from the SPECT and plasma data. In the three-compartment model, the first compartment consists of unmetabolized radioligand (i.e., [
123I]iomazenil) in plasma that is available for uptake into the brain, the second compartment is free and nonspecifically bound radioligand, and the third compartment is radioligand specifically bound to the receptor. Brain tissue activity as determined from the SPECT image includes radioligand specifically bound to receptor, nonspecifically bound to brain tissue, and free radioligand in brain tissue. However, the primary contribution to the image is from radioligand specifically bound to receptor
(32), and the contributions from nonspecific binding and free radioligand in brain tissue are assumed to be negligible. On the basis of this assumption, distribution volume is equal to the ratio of the concentration of bound radiotracer in brain to the concentration of free nonprotein bound radiotracer in plasma. This ratio of bound to free, also described as the binding potential, is equal to the ratio of receptor number (B
max) to affinity (K
D), and is also equal to (K
1 * K
3)/(k
2 * k
4 * f
1), where K
1 = rate of transfer from blood to brain; k
2 = rate of transfer from brain to blood; K
3 = rate of transfer from nonspecific or unbound to radioligand specifically bound to receptor; k
4 = rate of transfer from specifically bound to nonspecifically bound, and; f
1 = the fraction of free parent compound in plasma that is non-protein-bound and available for uptake into the brain. Data were analyzed by using both protein- and non-protein-bound plasma concentrations of radioligand. Identical results were obtained using both methods—data presented are based on protein-bound plasma concentrations of radioligand.
SPECT images in which values for individual voxels (or picture elements) within the brain corresponded to distribution volume values were then constructed by dividing values for radioactivity in each voxel by the plasma concentration of unmetabolized radioligand determined for that particular subject. These SPECT distribution volume images were stripped of extracranial artifact and coregistered to MR images. Coregistered MR images were used to determine normalization parameters for transformation of SPECT images into a common anatomical space. This method takes advantage of the superior delineation of anatomical detail by MR imaging in comparison to SPECT. Images were then smoothed by using a three-dimensional Gaussian filter to 16 mm full width at half maximum.
Statistical parametric mapping was used to compare distribution volumes (the measure of regional benzodiazepine receptor binding) in patients with PTSD versus comparison subjects
(33). Analyses were performed by using analysis of covariance with global benzodiazepine receptor binding considered as a confounding covariate. Statistical parametric mapping allows a statistical comparison between patients and comparison subjects on a voxel-by-voxel basis (voxels are the individual picture elements of a brain image, which correspond to a discrete location within the brain). With this method there is a statistical value (e.g., t statistic) for the difference between groups assigned to every voxel in the brain. Values for t statistic in individual voxels were transformed into z scores and displayed as an image made up of multiple voxels (or picture elements) that corresponded to individual anatomical areas of the brain, which demonstrated areas of statistically significant differences between the groups. Statistical parametric mapping analysis provides a global survey of areas of statistically significant difference throughout the brain. This approach has the inherent risk of falsely finding a significant difference due to multiple comparisons. To limit the risks of multiple comparisons, we used research findings from animal studies to identify a priori brain areas that would be expected to show a decrease in benzodiazepine receptor binding in PTSD. Regions selected included the hippocampus and prefrontal cortex, since two or more studies of the effects of stress on benzodiazepine receptor binding in animals have found decreased binding in these areas. Several studies have reported a decrease in the cerebral cortex, although studies that looked specifically at the occipital cortex found no change with stress. Since the cerebral cortex is nonspecific (and also includes the frontal cortex), the prefrontal cortex was identified as the a priori region of interest. This method of identifying a priori regions of interest has limitations, such as the fact that findings in rats may not be applicable to humans, the human brain is obviously more developed, and other brain areas may be affected in PTSD. However, in the absence of any literature on benzodiazepine receptor binding in PTSD, animal studies were used to develop hypotheses. Areas of decreased benzodiazepine receptor binding in PTSD within these hypothesized regions were identified by using a threshold of p<0.001 (z score >3.2). Other brain areas of significant difference outside of the hypothesized regions (if present) were identified for hypothesis generation for future studies. Location of areas of significant difference were identified as the distance from the anterior commissure in mm, with x, y, and z coordinates, by using the standard Talairach coordinate space
(34) with statistical maps displayed in radiological convention (patient’s left is image right). Pearson correlations were used to examine the correlation between benzodiazepine receptor binding in the prefrontal cortex and panic symptoms (measured with the clinician-rated Panic Attack Symptom Scale), anxiety symptoms (measured with the Hamilton anxiety scale), and general psychiatric symptoms (measured with the BPRS) at the time of scan.
Discussion
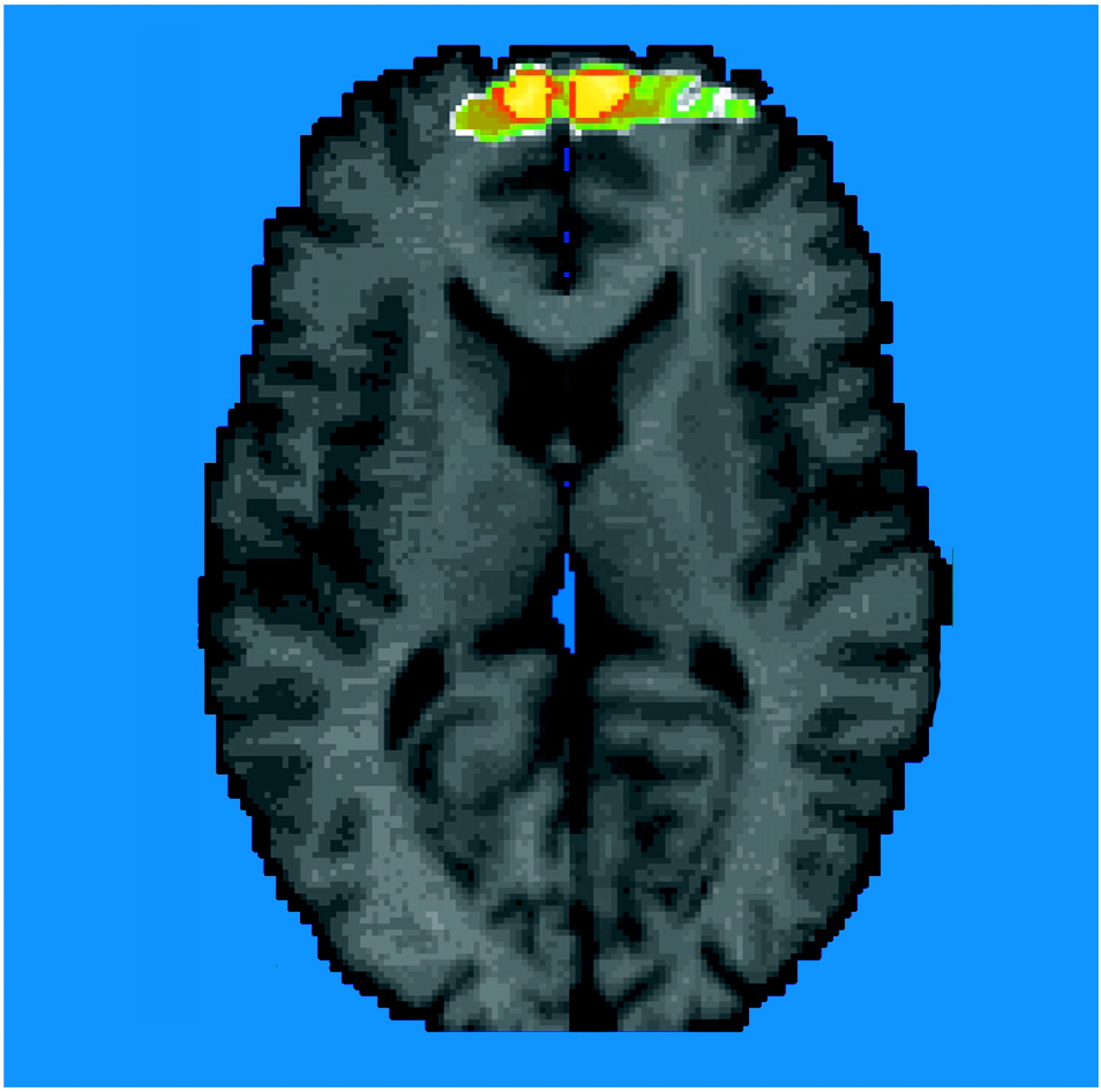
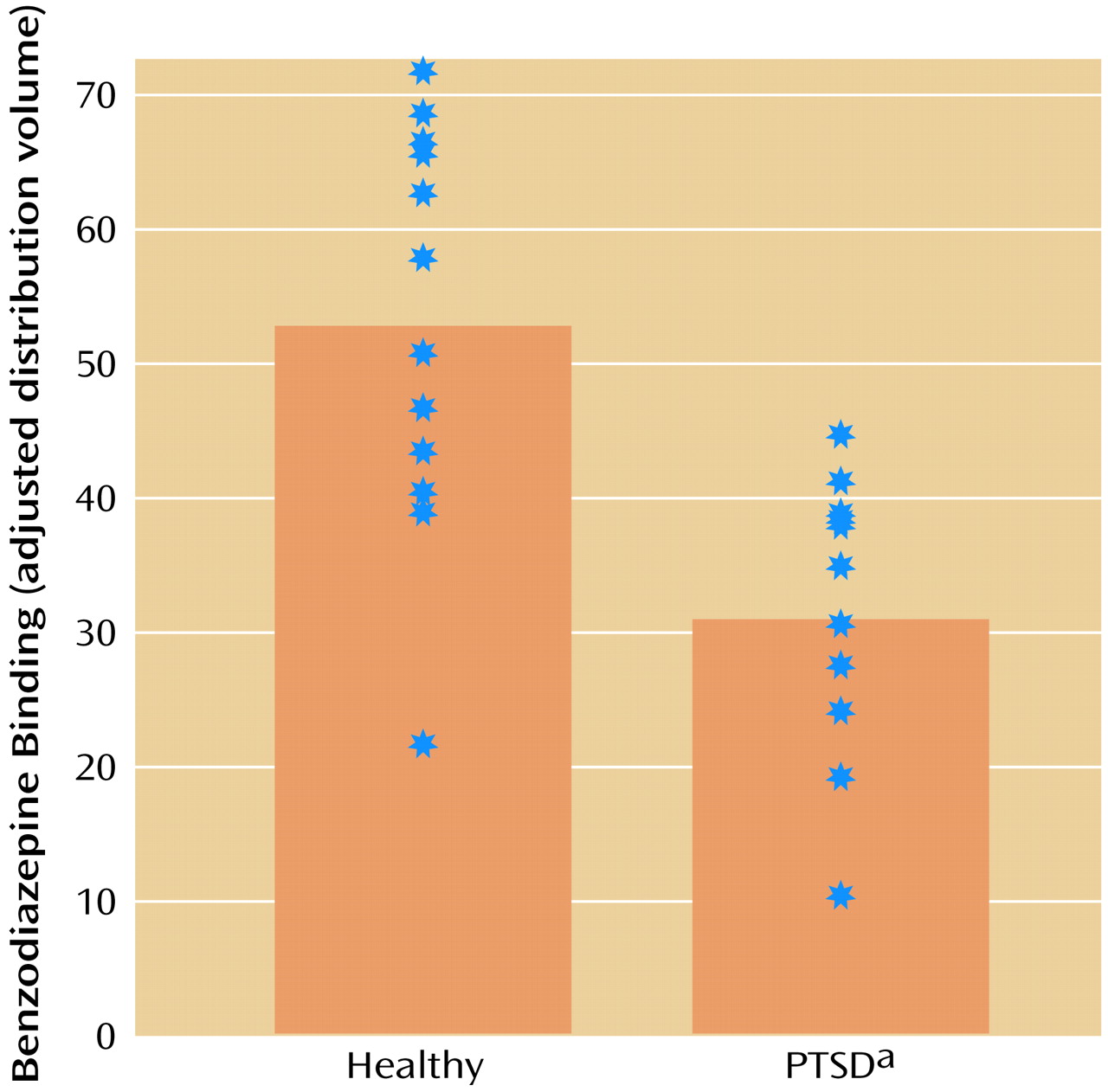
SPECT imaging of [123]iomazenil binding showed that PTSD patients had 41% lower distribution volumes (the measure of benzodiazepine receptor binding) in the prefrontal cortex (Brodmann’s area 9) relative to comparison subjects. There were no other areas of decreased binding or increased binding in PTSD patients relative to comparison subjects in this study.
These findings are consistent with preclinical studies that have shown a decrease in benzodiazepine receptor binding in the frontal cortex following stress. The findings could be related to a down-regulation of benzodiazepine receptor binding following exposure to the stress of war. Other possible explanations are that stress results in changes in receptor affinity, changes in an endogenous benzodiazepine ligand (the existence of which is controversial), or stress-related alterations in GABAergic transmission or neurosteroids that affect benzodiazepine receptor binding. An alternative explanation is that a preexisting low level of benzodiazepine receptor binding in the prefrontal cortex increases the risk for the development of PTSD following exposure to the stress of combat. Although a loss of neurons in prefrontal cortex in PTSD could explain a decrease in binding, the results are not explainable by differences in blood flow, since the equilibrium method eliminates differences in flow and tracer delivery from influencing the outcome measure of receptor binding.
The prefrontal cortex mediates several cognitive and behavioral processes that are potentially relevant to PTSD
(35). The medial prefrontal cortex has been implicated in the inhibition of cognition (which may relate to intrusive memories in PTSD) and emotional and social behavior
(36,
37). Positron emission tomography studies are consistent with medial prefrontal cortex dysfunction in PTSD, which provides a possible neural correlate of dysregulation of emotional and social behavior in PTSD
(38–
40). In one study of abuse-related PTSD
(39), traumatic reminders resulted in increased activation in Brodmann’s area 9 of the prefrontal cortex, the specific area of the prefrontal cortex implicated in this study. These findings point to the importance that alterations in the medial prefrontal cortex play in PTSD, although the role of specific subregions of this area in symptoms of this disorder are not yet clear.
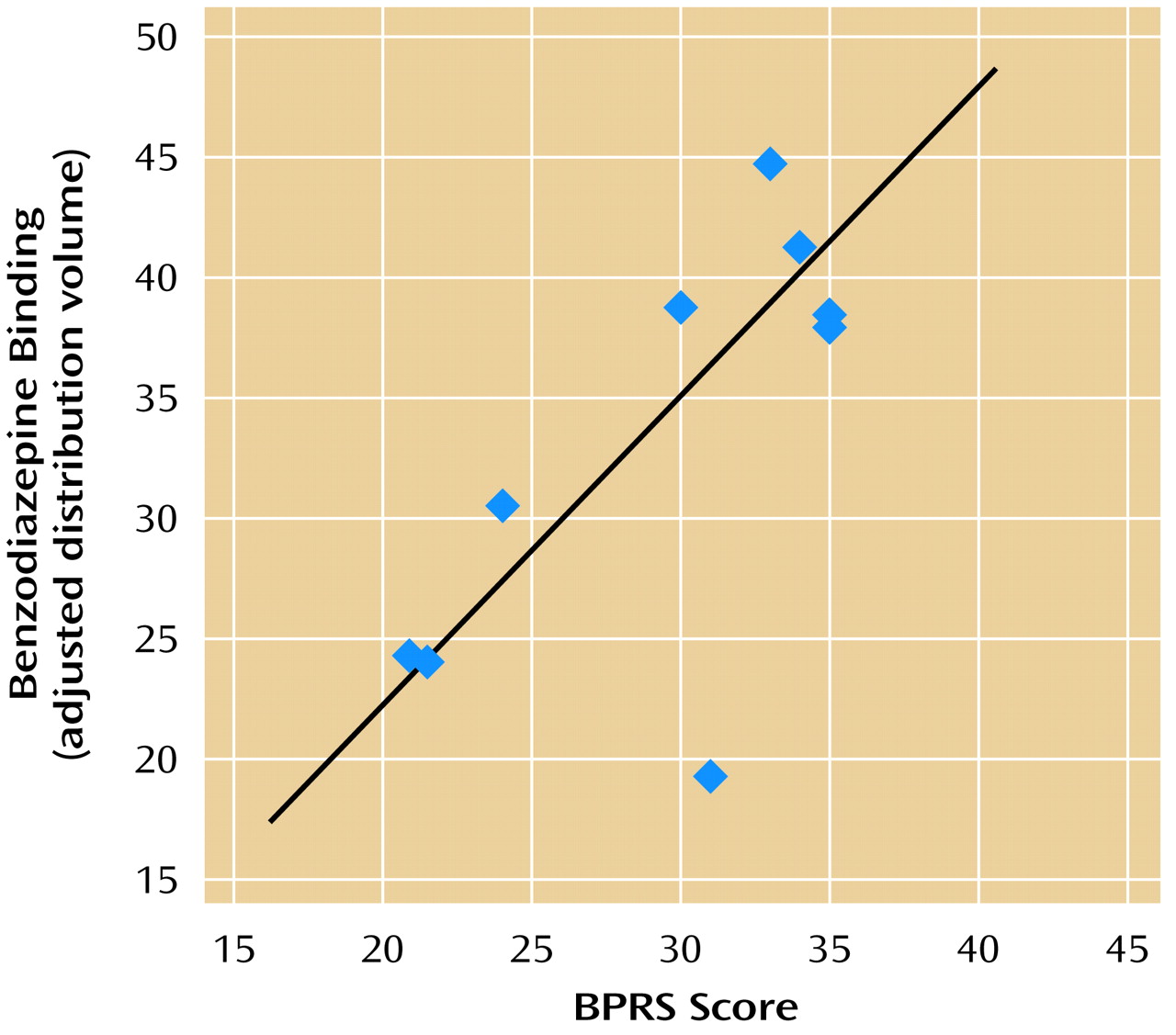
There was a positive correlation between psychiatric symptom level at the time of the scan as measured with the BPRS and decreased benzodiazepine receptor binding in the prefrontal cortex (Brodmann’s area 9). We hypothesized a priori a negative correlation between symptom level and prefrontal binding (not a positive correlation as our data showed). One interpretation of the findings is that chronic stress is associated with benzodiazepine receptor binding reduction, whereas acute increases in psychiatric symptom states are associated with increased benzodiazepine receptor binding in the prefrontal cortex (possibly related to down-regulation of receptors in response to repeated episodes of state anxiety). These findings require replication.
Several other limitations of this study are worth mentioning. There are assumptions inherent in the SPECT equilibrium technique for quantitation of neuroreceptor binding that need to be considered in the interpretation of the results. The method assumes that there is perfect equilibration between free activity in plasma and non-receptor-bound activity in the brain. The method also assumes the nonspecific binding in the brain is negligible (given the 20:1 ratio of specific to nonspecific binding of this radioligand); however, as much as 13% of radioligand is nondisplaceable
(25). Changes in the outcome measure (distribution volume) may be related to changes in neuroreceptor number or affinity. However, other factors, such as changes in endogenous ligands competing for the receptor site, could influence this parameter. Violations of any of these assumptions may influence quantitation of neuroreceptor binding, although this should not introduce a systematic confound in a study like the current one. It should be noted that there are advantages to the equilibrium technique, primarily because of the greater feasibility related to the fact that arterial sampling is not required. PTSD patients had other comorbid conditions, which is a typical finding in this disorder. Two of the PTSD patients had a past history of alcohol dependence, which may have contributed to the findings of decreased benzodiazepine receptor binding in the frontal cortex. There were high rates of anxiety disorders (particularly panic disorder) and depression, as previously reported. In all cases, the onset of PTSD preceded the onset of anxiety disorders and depression by several years or more. In a prior study, we reported decreased left hippocampal benzodiazepine receptor binding in panic disorder
(41), with no differences in the prefrontal cortex between patients and comparison subjects, although a correlation was found in a similar region of the prefrontal cortex in the panic patients. In our clinical judgment, symptoms that technically met criteria for panic disorder in the PTSD group were related to trauma exposure and were secondary to PTSD, and the patients in the two studies had very different clinical presentations. Therefore, it would be difficult to attribute findings of the current study to panic disorder. No studies have examined benzodiazepine receptor binding in depression. Again, however, patients in this study all had depression that followed the onset of PTSD and was clinically secondary as a diagnosis to PTSD. The current study did not include a combat-exposed non-PTSD control group. Therefore, it is possible that the findings are related to a nonspecific effect of extreme stress that is not specific to PTSD. Future studies are needed that use such a control. In the current study, we selected patients who were free of benzodiazepine usage or other substances as determined by toxicology screen and self-report. Although to the best of our knowledge patients had been free of benzodiazepines in the preceding 6 months, more remote histories were difficult to obtain.