Several lines of evidence, including in vivo human imaging results and postmortem tissue findings, suggest abnormalities of human hippocampal structure and function in schizophrenia. In vivo, hippocampal size in persons with schizophrenia is reduced bilaterally, albeit mildly, especially in anterior regions
(1–
5). Structural and histologic abnormalities in postmortem hippocampal tissue from affected persons have been repeatedly reported
(6–
11), although some of these findings have not been consistently replicated
(12). Moreover, in vivo functional studies of persons with schizophrenia have directly demonstrated an alteration in neuronal activity in the limbic system and/or parahippocampal gyrus, as measured by positron emission tomography and [
18F]fluorodeoxyglucose or regional cerebral blood flow techniques
(13–
17). In addition, considerable evidence of compromised cognitive function, especially in short-term memory and attention, exists in schizophrenia
(18–
20); these dysfunctions may represent the behavioral correlates of hippocampal pathology.
Studies from our laboratory using an in vivo animal preparation of psychosis with phencyclidine (PCP) support this focus. Because PCP and its congener ketamine induce psychotic-like phenomena in normal persons
(21–
23) and psychosis exacerbation in schizophrenia
(21,
24), PCP’s actions in animals have been studied to shed light on the mechanisms of psychosis, and even of schizophrenia, in humans
(25). Changes in neuronal activity and immediate early gene activation/repression in rat brain after PCP occur predominantly in limbic regions, including hippocampus
(26,
27). Moreover, an increase in
N-methyl-
d-aspartic acid (NMDA)-sensitive glutamate receptor binding occurs exclusively in rat hippocampus 12–24 hours after PCP administration
(28,
29). The selective PCP agonist MK801 also demonstrates these actions. One parsimonious explanation for these PCP-induced neurochemical changes in rat hippocampus is that PCP administration causes inhibition at the NMDA-sensitive glutamate receptor in the trisynaptic perforant pathway, and the effect is additive within the hippocampal projection fields and becomes greatest in the Schaffer collateral terminal field of CA1. This inhibition could result in a functionally significant reduction in glutamatergic output from the hippocampal cortex to other limbic regions, including but not limited to an effect on the anterior cingulate cortex. We have speculated that this reduction in excitatory output from hippocampus and reduced afferent cingulate stimulation is associated with psychosis across psychotic diagnoses, whether the psychotic symptoms are induced by PCP or associated with schizophrenic psychosis
(25).
Discussion
The hippocampus has been a consistent focus of biologic study in schizophrenia, yielding several replicable observations of abnormality. This emphasis has been encouraged by observations of behavioral disruptions in the illness that suggest hippocampal dysfunction
(40,
41). The behavioral actions of PCP in healthy humans and in schizophrenia have implicated the blockade of glutamate transmission at the NMDA receptor in psychosis
(22,
24,
25,
30,
42). Moreover, specific anatomic data exist (reviewed below) that support hippocampal pathology related to glutamate in the illness.
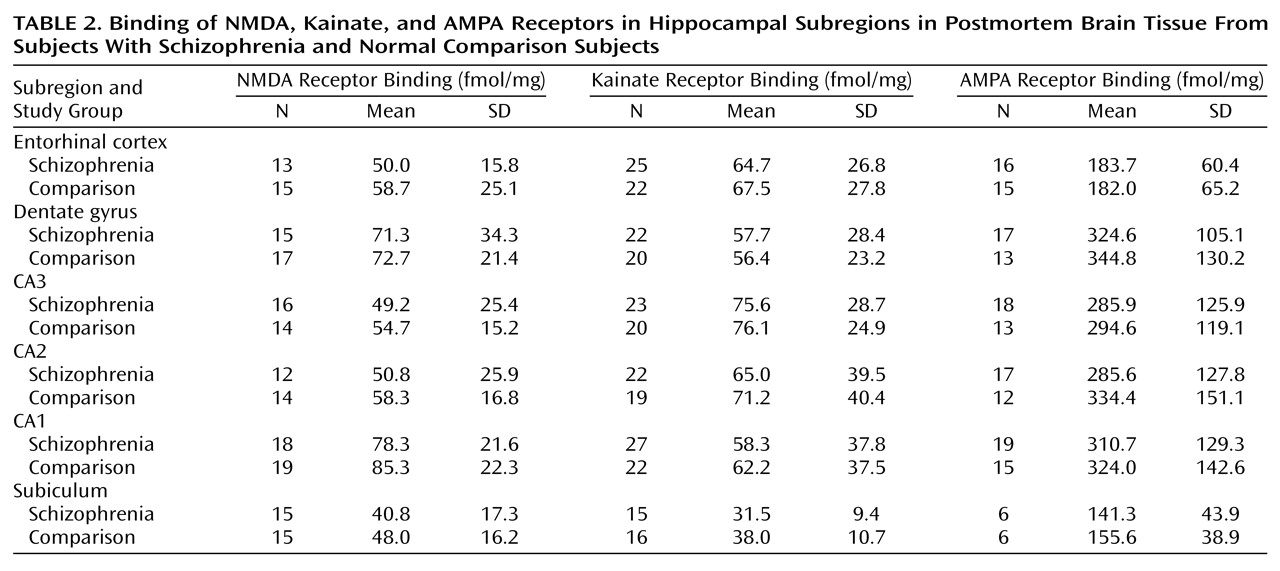
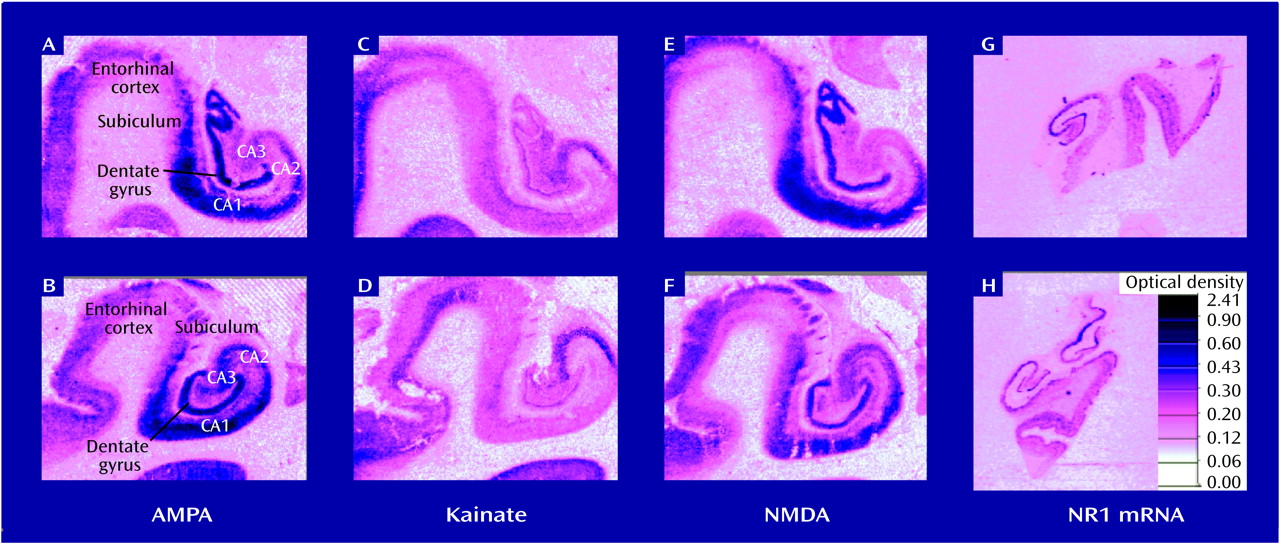
Several alterations in neurochemical measures of glutamatergic and related transmitter function in the postmortem hippocampus and/or the related dorsoventral temporal cortex in schizophrenia have been identified. Because the glutamate system has several different receptor families, receptor configurations, transmitters/modulators, and modulating receptor sites, the task of focusing a search for a pathologic site involving glutamate neurotransmission has been challenging. Although there appears to be no difference in the density of hippocampal NMDA glutamate receptors in schizophrenia and in normal tissue
(39,
43,
44), a lower level of kainate binding, particularly in CA2, has been found
(38,
39,
45), but not consistently
(46). Lower levels of non-NMDA receptor binding
(39) and lower concentrations of non-NMDA receptor mRNA
(47) have both been reported in CA3 in schizophrenia. Moreover, lower levels of the glutamate receptor subunit mRNAs for GluR2, KA2 and GluR6 (kainate and AMPA subunits) have been found selectively in hippocampus in schizophrenia
(48), and a higher flip/flop isoform ratio has been identified for AMPA receptors
(49). However, in one study, the level of GluR1–GluR7 mRNA subunits appeared no different in schizophrenia than in normal tissue in all brain regions evaluated
(50), whereas another study found a lower level of GluR1 mRNA in the parahippocampal gyrus and a lower level of GluR2 mRNA in several hippocampal subregions in schizophrenia
(51). These results suggest possible, but inconsistently found, differences in the kainate and AMPA receptor complexes in schizophrenia.
A higher level of binding to NR1/NR2B receptors has been reported in superior temporal cortex in schizophrenia
(52). The level of aspartate binding, putatively to presynaptic elements, has been reported to be either higher
(46) or no different
(45) in temporal cortex in schizophrenia than in normal tissue. Reports of lower levels of hippocampal synaptophysin are consistent with a lower density of synapses in hippocampus in schizophrenia
(53). Some evidence supports higher glutamate concentrations within temporal cortex, but not in hippocampus, in schizophrenia
(54). Release of glutamate and release of γ-aminobutyric acid (GABA) may be lower in schizophrenia tissue
(55). Differences in GABA
A receptor density, in GABA release, and in glutamate-related transmitters and their enzymes in hippocampus have been reported in the illness
(30,
45,
56). Often, more pronounced or significant differences have been found within the left compared to the right hemisphere of the temporal cortex
(57). Although these findings taken together do not support a single, simple interpretation of glutamatergic involvement in schizophrenia, they do indicate potential instability in ionotropic glutamate transmission in hippocampus in this illness. The data reported here fit into a growing published literature on the complexity of glutamatergic transmission in brain, its distinctive regional characteristics, and our still evolving understanding of the primary elements of the system
(58).
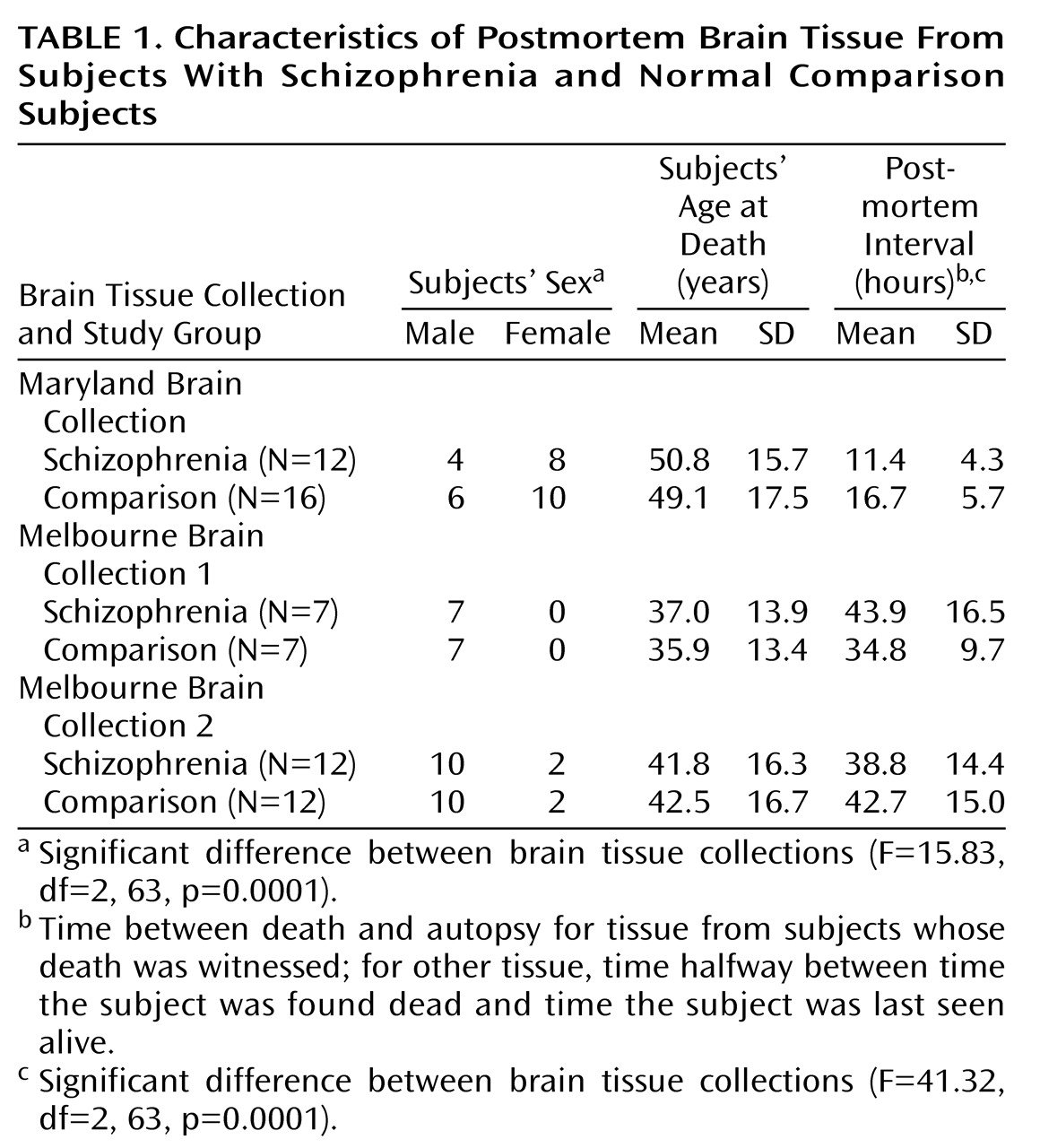
In the study reported here, the hippocampal block dissections were not entirely standardized with respect to their position along the anterior-posterior axis; thus, the pathologic findings could not be localized along the long axis of the hippocampus with any precision. On the basis of segregated afferent and efferent pathways and the now suggested anterior-posterior hippocampal differences in schizophrenia
(59), it has become apparent that this localization may be important. Consequently, we have begun a new examination of the whole hippocampus in schizophrenia using a specialized tissue collection technique to check if these reported abnormalities are regionally localized within hippocampus.
The hypothesis underlying the study reported here was based on the possibility that PCP-induced psychosis and schizophrenic psychosis might share a common biologic mechanism. The mechanism of PCP-induced psychosis, inferred from animal studies done in this laboratory
(27,
28), is reduced glutamatergic transmission at the NMDA receptor within the trisynaptic hippocampal glutamatergic pathway
(25). These findings suggest the simplistic idea that schizophrenia tissue would evidence the same up-regulation of the NMDA receptor (as a marker of glutamatergic transmission at those hippocampal synapses) as the rat PCP preparation
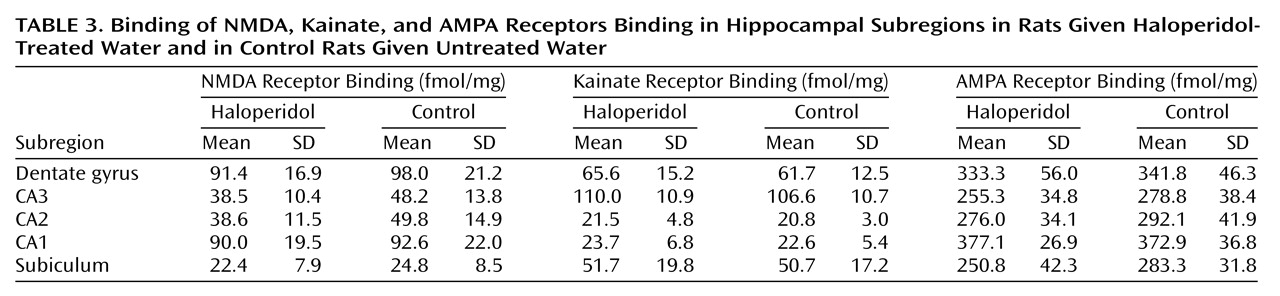
(28). The rat studies of chronic haloperidol administration showed no difference in these parameters with antidopaminergic actions, suggesting that findings for NMDA receptors could be distinguished from the effects of chronic drug treatment.
The initial hypothesis, that we would find NMDA receptor up-regulation in hippocampus, particularly in CA1
or CA3, secondary to an internal disruption of hippocampal glutamatergic transmission, was not confirmed by this study. Using levels of receptor density as an index of an alteration in glutamate mediated transmission in hippocampus, the results suggest no differences between the schizophrenia tissue and the normal comparison tissue. These results are consistent with reports from other laboratories
(39,
43,
44).
Subsequent analysis of the expression of NMDA receptor subunits was indicated to fully examine the a priori hypothesis. The NR1
receptor subunit of the multimeric NMDA receptor is the critical subunit for the ionophore function of gating calcium ions. The NR1
subunit has multiple isoforms that have distinct pharmacologic properties
(60); consequently considerable diversity exists among brain NMDA receptors. The NR2 family of NMDA subunits, which are also found in hippocampus, have four members: NR2A, NR2B, NR2C, and NR2D. The NMDA receptor on hippocampal pyramidal cells is thought to be composed of NR1, NR2A, and NR2B subunits, with the NR2A and NR2B subunits present in only moderate concentrations. The NR2C and NR2D subunits are expressed at the lowest levels in hippocampus and are thought to combine with the NR1 subunit located preferentially on the hippocampal interneurons. Experimentally constituted receptors, lacking the NR1 subunit, fail to generate a functioning NMDA ionophore that can gate ionic calcium flow
(36). NMDA receptors composed of NR1, NR2A, and NR2B subunits demonstrate significant pharmacologic, functional, and anatomic differences from NMDA receptors composed of NR1, NR2C, and NR2D subunits
(60). For example, in recombinant NMDA receptor studies in vitro, the NR1-NR2A-NR2B receptors increased the elevation of calcium that is modulated by protein kinase C and mediated by NMDA receptors, whereas the NR1-NR2C-NR2D receptors decreased this intracellular calcium measure
(62). In this paper, we have evaluated the subunit message that purportedly directly modulates hippocampal pyramidal neuronal activity, i.e., the mRNA of the NR1, NR2A, and NR2B subunits.
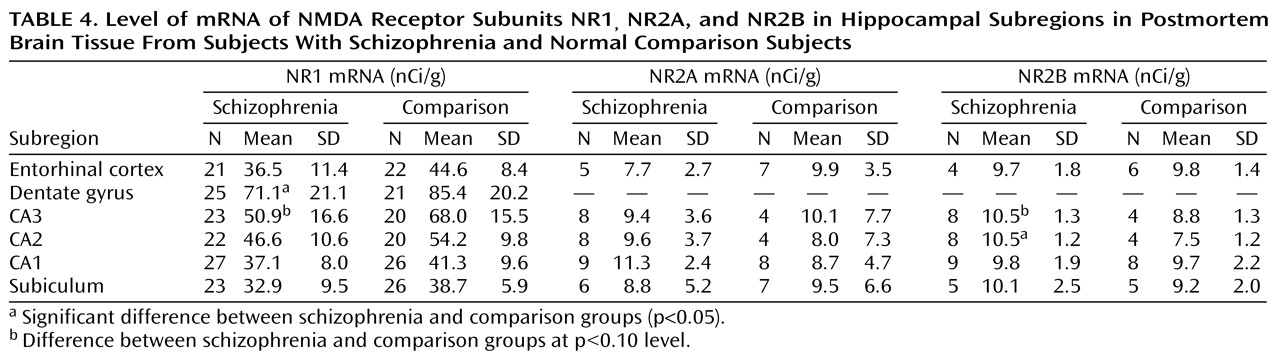
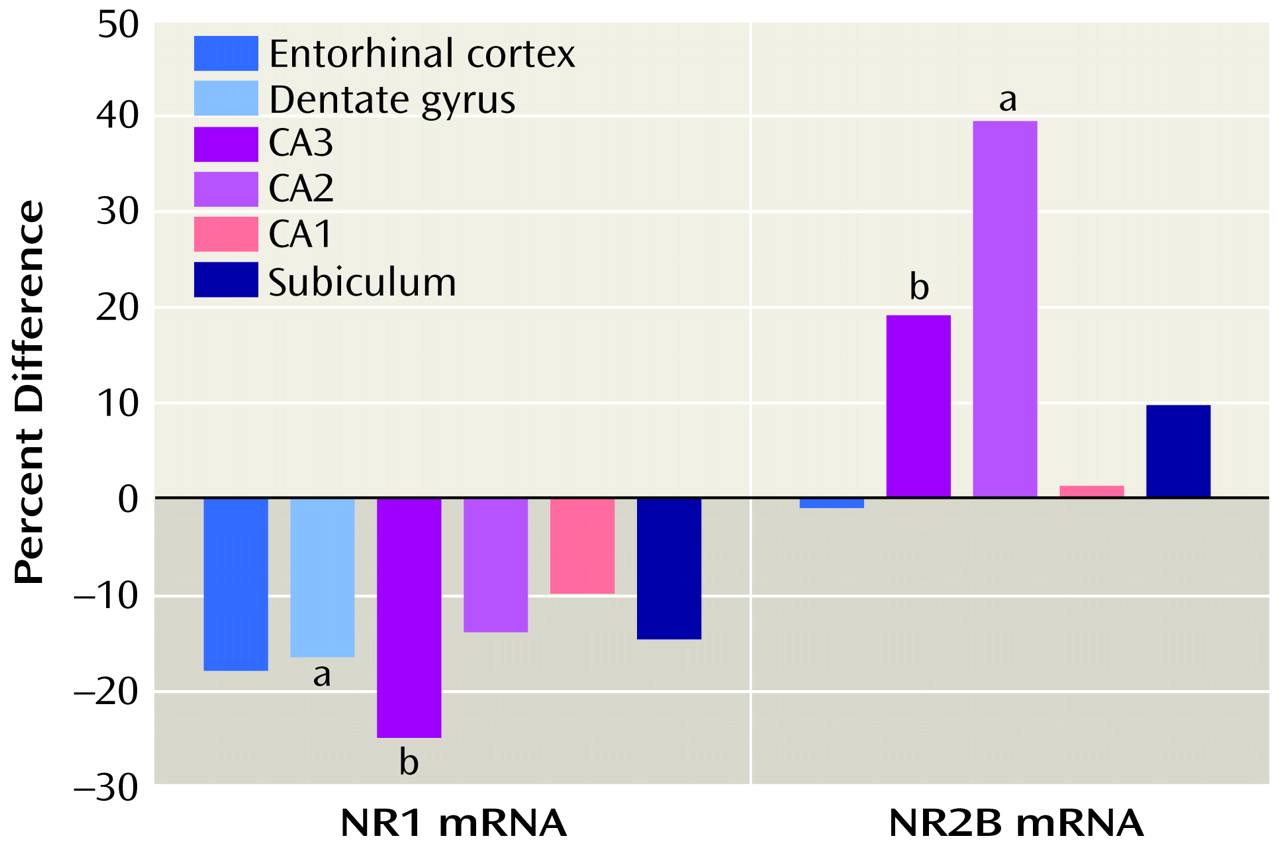
A lower level of NR1 expression and a higher level of NR2A or NR2B expression, as reported here, are consistent with the idea that the composition of at least some of the hippocampal NMDA receptors—those that reside on pyramidal neurons—may be abnormal in schizophrenia. A linkage analysis of schizophrenia in an African tribal family suggested that alterations in the NMDA receptor may provide a genetic predisposition to the illness
(63), although this observation needs further replication. Molecular techniques have provided mouse models of regionally reduced NR1-composed NMDA receptors using antisense
(64), adenovirus
(65), or customized recombination systems
(66). These mouse preparations show behavioral alterations in the whole animal
(67). Such animal models may provide critical preclinical systems for understanding the biologic consequences of lower levels of NR1. Indeed, transgenic mice with a lower than normal expression of the NR1 NMDA receptor subunit have some characteristics similar to those observed in pharmacologically induced animal models of schizophrenia
(68).
An NMDA receptor lacking the NR1 subunit would be unable to gate calcium and thus would not be fully functional, but it could demonstrate full ligand binding. One might tentatively suggest, based on this evidence, that several subregions of the hippocampus may have a lower number of functioning NMDA receptors in schizophrenia, compared with normal tissue, and may thus be “hypoglutamatergic.” This altered receptor mechanism would reduce NMDA-mediated neural transmission not through receptor blockade but by abnormal receptor composition and consequently abnormal function. The outcome of this difference in receptor composition would be reduced transmission at the NMDA receptor. Perhaps any mechanism that compromises hippocampal NMDA-sensitive glutamate-mediated neurotransmission in an analogous fashion would produce psychosis. These speculations would be further supported by the demonstration of lower levels of NR1 protein and higher levels of NR2A and NR2B protein in hippocampal subregions in schizophrenia and by documentation of NR2C and NR2D levels, efforts that are now ongoing in our laboratory.
These early studies suggest an abnormality of the composition of the NMDA-sensitive glutamate receptor in hippocampus in schizophrenia. Preliminary data from other laboratories show NMDA subunit alterations in thalamus
(69) and in superior temporal cortex
(52) in schizophrenia, suggesting that such differences in subunits might not be limited to hippocampus. Moreover, higher levels of NR2B subunit protein in hippocampus in schizophrenia has recently been reported from another laboratory, suggesting that higher levels of NR2B mRNA will translate into higher levels of NR2B protein
(70). Although the interpretation of the data remains tentative, it is consistent with considerable evidence documenting glutamate-related abnormalities of limbic cortex and pathology in hippocampus in schizophrenia.