Proteins are the final expression products of genes, the functional translation of the genomic information, conveyed by mRNA. Some experimentalists ask, Why measure mRNA at all, if it is only an intermediate on the way to production of the functional protein products? The most important reasons are of a practical nature: nucleic acid analyses are relatively easy, are very sensitive by virtue of the possibility of amplification, and can be readily automated. However, all of this does not diminish the need for protein information, as well as RNA expression, to fully characterize the information content of cells, tissues, or body fluids. Moreover, only the characterization of the proteins themselves can reveal posttranslational modifications (e.g., phosphorylation, sulfation, glycosylation) and give insight into protein-protein interactions and subcellular localization, thus providing clues about function. The large-scale identification of proteins contained in a tissue is often referred to as “proteomics.” It is frequently carried out by using two-dimensional gel electrophoresis as a separation method and followed up with mass spectroscopy for the identification and characterization of proteins of interest. Descriptively, the target tissue is harvested and the proteins solubilized. The crude protein mixture is then applied to a gel and separated according to electric charge along the first dimension followed by a separation according to size along a second dimension, perpendicular to the first one. The resulting proteome image (illustrated in the
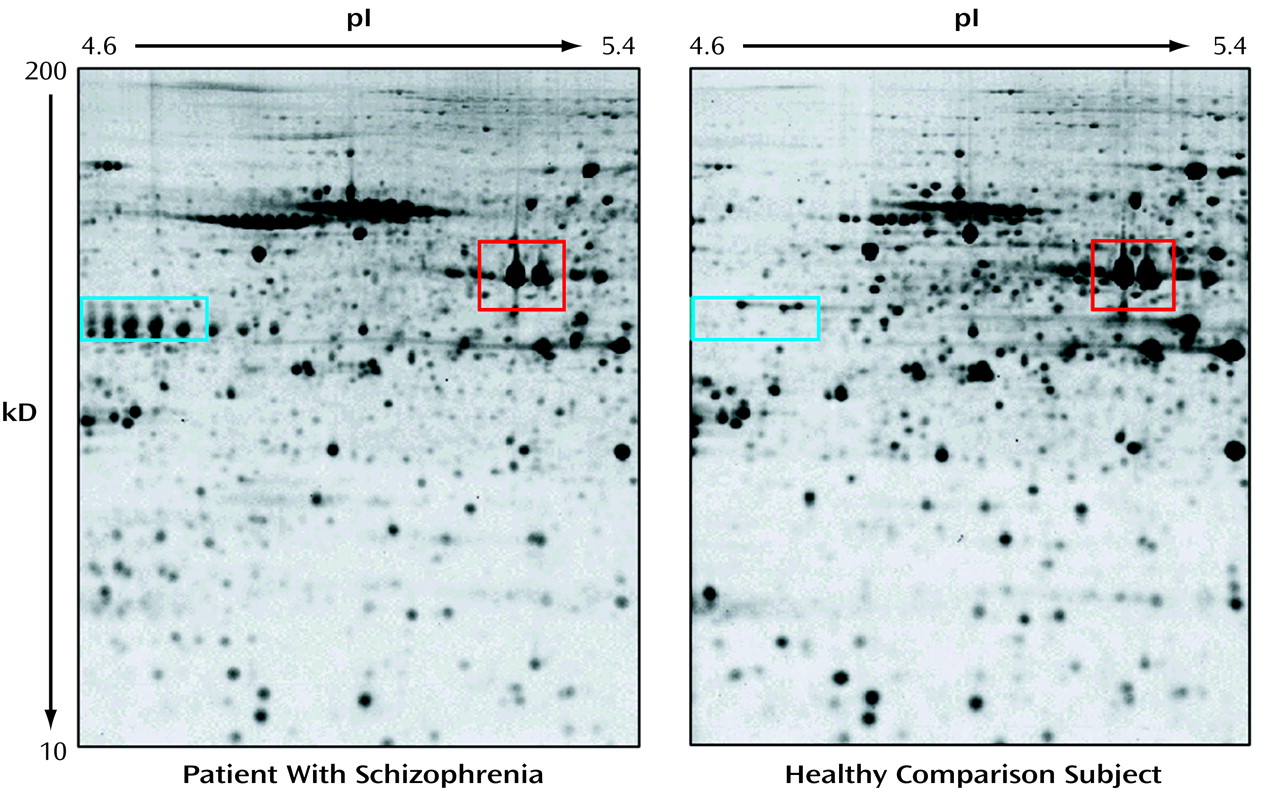
figure) is a complex array of spots, each of which represents an individual protein species. Analysis of the proteome image requires specialized computer programs that can match spot patterns across the different images, thus allowing for a comparison and statistical evaluation of protein profiles between tissues and across samples. Mass spectroscopy of an excised spot is the technique used to identify and characterize the protein. The mass spectroscopic identification of gel-separated proteins has been a highly important breakthrough in proteomics in the 1990s because it extends protein analysis far beyond the mere display of tissue protein patterns. The experimental comparison of the protein content from two different tissues (e.g., diseased versus healthy tissue) can directly answer the question of what protein differences characterize a specific tissue in a brain disease. This is illustrated in the figure, which depicts a proteome image of a specific brain area of a patient with schizophrenia (left) compared with that of a healthy individual (right). Besides a large number of common features, such as the two actin isoforms in the red rectangle, there are also a number of obvious differences, e.g., the presence or absence of isoforms of the glial acidic fibrillary protein (blue rectangle). This illustrates the intricacy of working with human tissue, where most of these differences are attributable to natural variation between individuals. In the analysis of complex brain diseases such as schizophrenia, the challenge for proteomics is to provide methods and tools to filter out the disease-specific changes from polymorphisms that simply discriminate one individual from another.