Initiation of drug therapy soon after the onset of psychotic symptoms is associated with better medication response, less likelihood of relapse, and more favorable long-term outcome among patients with schizophrenia
(1). These findings raise the possibility that early pharmacological intervention arrests or attenuates deteriorative aspects of the schizophrenic disease process
(2). If so, even greater benefits may be achieved by initiating treatment during the prodromal phase of schizophrenia
(3). As a preliminary step toward testing this hypothesis, we conducted an open-label study to evaluate the safety and tolerability of short-term treatment with a low dose of an atypical antipsychotic drug—risperidone—among treatment-seeking adolescents with prodromal symptoms and a family history of schizophrenia. We evaluated change in symptoms and neurocognitive test performance from baseline to 8–12-week follow-up and compared the magnitude of change between the prodromal patients and a parallel group of first-episode schizophrenic patients.
Method
This was an investigator-initiated, Finnish-multicenter, open-label, phase-IIIb trial. The study protocol was reviewed and approved by the institutional review board of the Finnish National Agency for Medicines and by the institutional review boards of all participating psychiatric facilities. Before participation, all subjects were given study information by the treating psychiatrist and signed informed consent forms. For subjects under 18 years of age, the subjects’ parents or legal guardians also signed informed consent forms.
Prodromal subjects were treatment-seeking, neuroleptic-naive outpatients, aged between 15 and 20 years, who met DSM-IV criteria for schizotypal, paranoid, or schizoid personality disorder but not for schizophrenia, schizoaffective disorder, schizophreniform disorder, or a psychotic mood disorder and had at least one first- or second-degree relative with a DSM-IV diagnosis of schizophrenia. First-episode subjects were in- or outpatients, aged between 16 and 35 years, who met DSM-IV criteria for schizophrenia or schizophreniform disorder, with a maximum of 2 weeks of prior neuroleptic medication. Exclusion criteria for all subjects were existence of any other axis I disorder, alcohol or drug dependence, any severe drug allergy, any significant cardiac abnormality, any other serious medical illness, mental retardation, and history of suicide attempt or acute suicidal risk. Diagnoses were made by using the SCID interview
(4). Pregnant or lactating women and women of childbearing potential who were not using an effective means of contraception were also excluded.
The trial medication (risperidone) was administered once daily for 8 weeks to first-episode patients and for 12 weeks to prodromal patients. For both prodromal and first-episode patients, the treatment started at 1 mg/day. For prodromal subjects, the dose was increased after 2 weeks by 0.5 mg/day and after 4 weeks by 0.5 mg/day up to a maximum daily dose of 2 mg. For first-episode subjects, the dose was increased after 3 days by 1 mg/day and thereafter every week by 1 mg/day up to a maximum daily dose of 4 mg. For both groups, doses could be titrated downward at any time in the event of clinically significant treatment-emergent adverse events or if, in the treating psychiatrist’s judgment, a decrease was warranted. No antidepressants, mood stabilizers, or antipsychotics other than the study drug were allowed for the duration of the trial. For both groups, anticholinergic medication could be prescribed for emergent extrapyramidal symptoms whenever necessary for a maximum of 6 consecutive days. Withdrawal from the trial was at the discretion of the treating psychiatrist for safety or efficacy considerations.
For all subjects, a standard form was used to elicit and record information on demographic status and psychiatric and medical history. Symptom ratings and neurocognitive measures were obtained at baseline and at two subsequent follow-ups, which occurred at 4 and 8 weeks for first-episode patients and at 6 and 12 weeks for prodromal patients. For prodromal subjects, symptoms were rated by using the Child Behavior Checklist
(5); analyses used the average of the self- and parent-reported ratings (which did not differ significantly). For first-episode patients, symptoms were rated by the treating psychiatrist by use of the Positive and Negative Syndrome Scale
(6). All subjects were given the California Verbal Learning Test
(7). Adverse events were monitored continuously with a standard checklist completed at baseline and at 2-, 4-, 6-, 8-, and 12-week follow-ups.
Results
A total of 16 subjects were enrolled in the study. Of these, five were prodromal patients (mean age=15.6 years, SD=0.8; three boys and two girls), four of whom completed the trial, and 11 were first-episode patients (mean age=23.9 years, SD=5.5; eight men and three women), six of whom completed the trial. The mean dose of risperidone was 1.04 mg/day (SD=0.12) among prodromal patients and 1.83 mg/day (SD=0.50) among first-episode patients. Adverse events judged to be possibly or probably related to the treatment were noted in two prodromal patients (tremor/tic, exhaustion/anxiety) and three first-episode patients (sedation, dystonia, dry mouth/rhinitis); all of these were minor and resolved during the trial. Of the six patients who dropped out, only one (a first-episode patient) withdrew because of adverse events. No adverse reactions were observed in the one prodromal patient whose parents withdrew consent to participate.
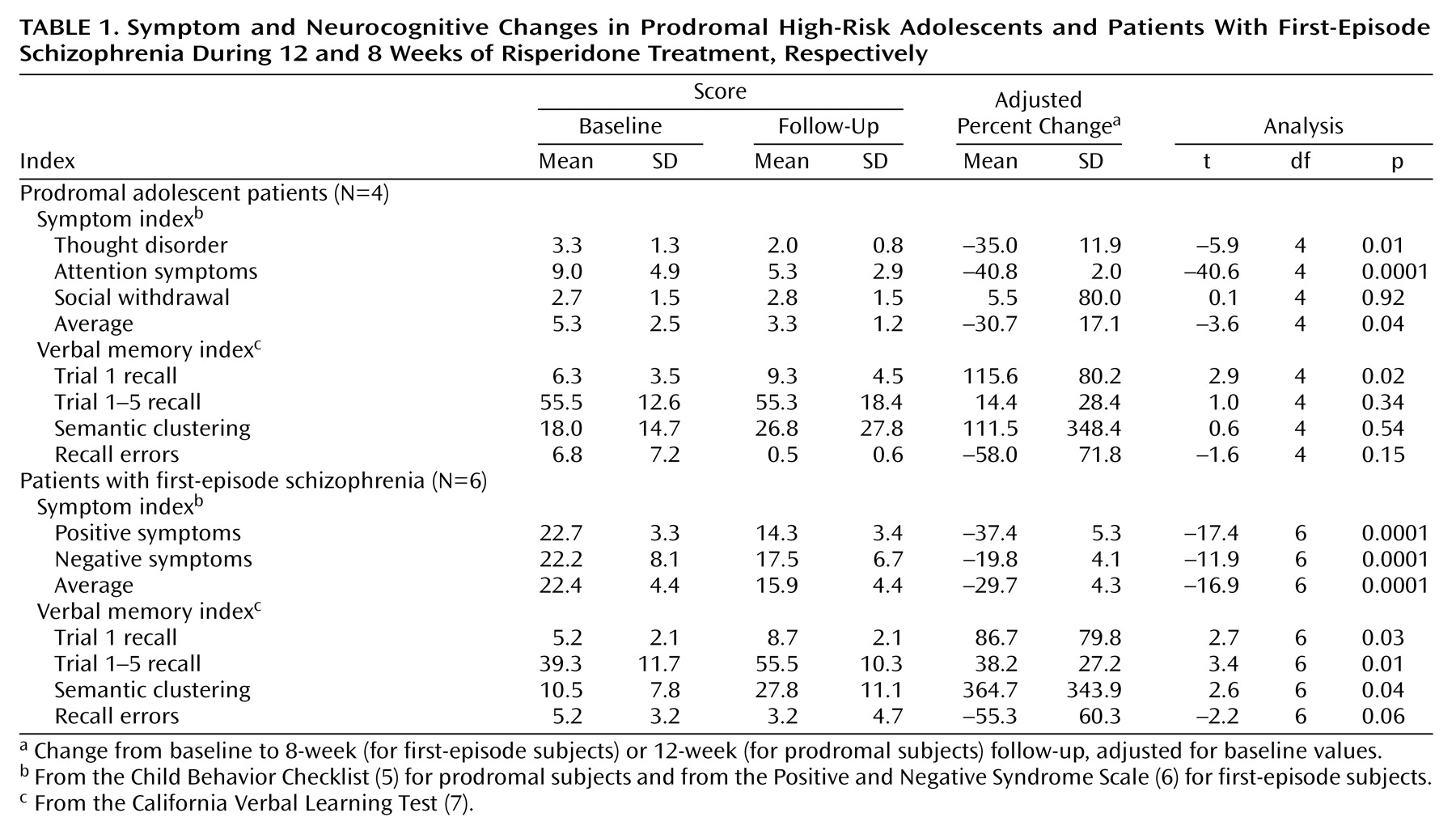
Table 1 gives information on symptom severity and neurocognitive functioning in the prodromal and first-episode groups. On average, prodromal patients’ Child Behavior Checklist scores declined significantly at the 12-week follow-up, compared to baseline scores, in terms of thought disorder, attention symptoms, and overall symptoms but not in terms of social functioning. All four prodromal subjects improved in terms of attention, and three of four improved in terms of thought disorder and overall symptoms. On average, first-episode patients’ Positive and Negative Syndrome Scale scores declined significantly overall and on the positive and negative subscales. All six first-episode patients improved in terms of positive and overall symptoms, and five of six improved in terms of negative symptoms.
Performance on the California Verbal Learning Test improved significantly from baseline to final follow-up in terms of trial 1 recall (mean change=98.2%, SD=92.8%) (t=3.3, df=10, p=0.008), overall recall on trials 1 to 5 (mean=28.7%, SD=27.8%) (t=3.3, df=10, p=0.009), semantic clustering during recall (mean=263.4%, SD=218.8%) (t=3.8, df=10, p=0.004), and recall errors (i.e., sum of intrusions and perseverations: mean=–54.6%, SD=12.2%) (t=–14.1, df=10, p=0.0001). The rate of improvement did not differ significantly between first-episode and prodromal subjects on any of these measures (t=0.6, df=9, p=0.59; t=–1.2, df=9, p=0.27; t=–1.1, df=9, p=0.31; t=–0.1, df=9, p=0.95, respectively). Significant change was observed on trial 1 recall among prodromal patients and on all measures but recall errors among first-episode patients.
Discussion
Short-term treatment with a low dose of risperidone was safe and may have been associated with improvement in behavioral and neurocognitive functioning in a small group of adolescent patients with prodromal symptoms of schizophrenia. Severity of thought and behavior disturbances declined by about 30%, and performance on indices of verbal learning improved by 30% to 100% during 8 to 12 weeks of treatment in both prodromal and first-episode patients. These changes were observed consistently across subjects and achieved statistical significance despite the small group sizes. It is encouraging that the response profile of prodromal subjects was similar to that of first-episode patients, among whom the efficacy of risperidone on clinical and cognitive functioning has previously been proven
(8). Nevertheless, given the small group size, lack of placebo control group, and failure to control for practice effects on the neurocognitive tests, the present findings are preliminary and should not be used to guide public health decisions regarding treatment of individuals with prodromal symptoms. Rather, the findings encourage larger, longer-term, randomized, placebo-controlled, double-blind clinical trials in this population.