Sensory gating abnormalities may index a genetic vulnerability for schizophrenia that reflects abnormal hippocampal function
(1). Adler et al. have demonstrated that normal sensory gating depends, in part, on nicotinic cholinergic modulation of inhibitory interneurons in a CA3 hippocampal circuit
(1). Consistent with this model, gating abnormalities in schizophrenia patients and their family members have been shown to improve after administration of nicotine
(2,
3) or donepezil
(4). Dopaminergic, GABAergic, noradrenergic, and serotonergic influences on sensory gating have also been documented
(1). Given the complexity of the possible pharmacological influences on sensory gating, it is important to evaluate antipsychotic medication effects on this putative vulnerability marker.
Conventional antipsychotic medications do not remediate sensory gating deficits for schizophrenia patients
(5). New-generation antipsychotic medications such as clozapine
(5–
7), olanzapine
(7), or risperidone
(8) may be associated with improvements in sensory gating. However, to our knowledge medication effects have never been investigated in the context of a randomized clinical trial. This study was designed to compare the effects of olanzapine and haloperidol on the P50 sensory gating index in treatment-resistant patients with schizophrenia.
Method
Patients participating in a 16-week, double-blind, parallel-group comparison of olanzapine and haloperidol were invited to enter the study. Patients met DSM-IV criteria for either schizophrenia or schizoaffective disorder. All participants provided written informed consent prior to study entry. Subjects with concurrent substance abuse or alcoholism, organic brain disorder, or mental retardation were excluded. The age range of the patient population was 18 to 60. Patients had to have demonstrated an inadequate treatment response to conventional antipsychotics, defined as 1) a history of two or more ineffective trials of at least two different classes of conventional antipsychotics, and 2) failure to respond to a prospective 4-week open-label trial of fluphenazine (mean dose=20 mg/day, range=10–30) and benztropine (4 mg/day). Patients who exhibited a 30% improvement in either the BPRS positive symptom score or the SANS total score were excluded from the study. Patients who experienced a relapse or were intolerant of fluphenazine were not eligible for participation in the study.
Patients who fulfilled treatment resistance criteria and were clinically stable were randomly assigned to either olanzapine or haloperidol. The optimal dose for both olanzapine and haloperidol was 15 mg/day, and both medications had dose ranges of 10–30 mg/day. Benztropine, 4 mg/day, was prescribed to patients randomly assigned to the haloperidol condition to minimize extrapyramidal symptoms. Patients assigned to olanzapine received placebo benztropine.
Sensory gating measures were obtained from each participant at baseline, while patients were receiving fluphenazine and benztropine, and 12 weeks after randomization. P50 event-related potentials were recorded with tin electrodes (Electro-Cap International, Eaton, Ohio) placed at midline locations (FPz, Fz, FCz, Cz, CPz, Pz, Oz) referenced to linked earlobes during a paired-click procedure (75-dB clicks, 150 click pairs, 10-second interpair interval, 0.5-second interclick interval). Eye movements were monitored with an electrode placed at the outer canthus of the right eye, which was referred to the right frontal pole. Neurophysiological signals were sampled at 1000 Hz with a Neuroscan Synamp amplifier (200 Hz low pass, 1.0 Hz high pass) to yield 500-msec epochs (100-msec prestimulus window). Signal averaging was conducted offline to permit the rejection of single trials that contained movement artifact (rejection criterion of ±75 μV). P50 amplitude and latency measures were made on bandpass filtered (10–100 Hz, 12 dB) averages obtained from Cz. For the first click, P50 was defined as the largest positive-going wave occurring within 35–70 msec poststimulus. Amplitude was measured from the trough of the preceding wave to the P50 peak. The P50 latency window for the second click was set to within ±10 msec of the P50 latency for the first click. P50 amplitude measures were used to construct sensory gating ratios (P50 amplitude following the second click divided by the P50 amplitude following the first click).
Repeated-measures analysis of variance was used to evaluate the effects of time (baseline, endpoint) and medication (olanzapine, haloperidol) and their interaction. Chi-square was used to compare normal suppression rates at baseline and endpoint.
Results
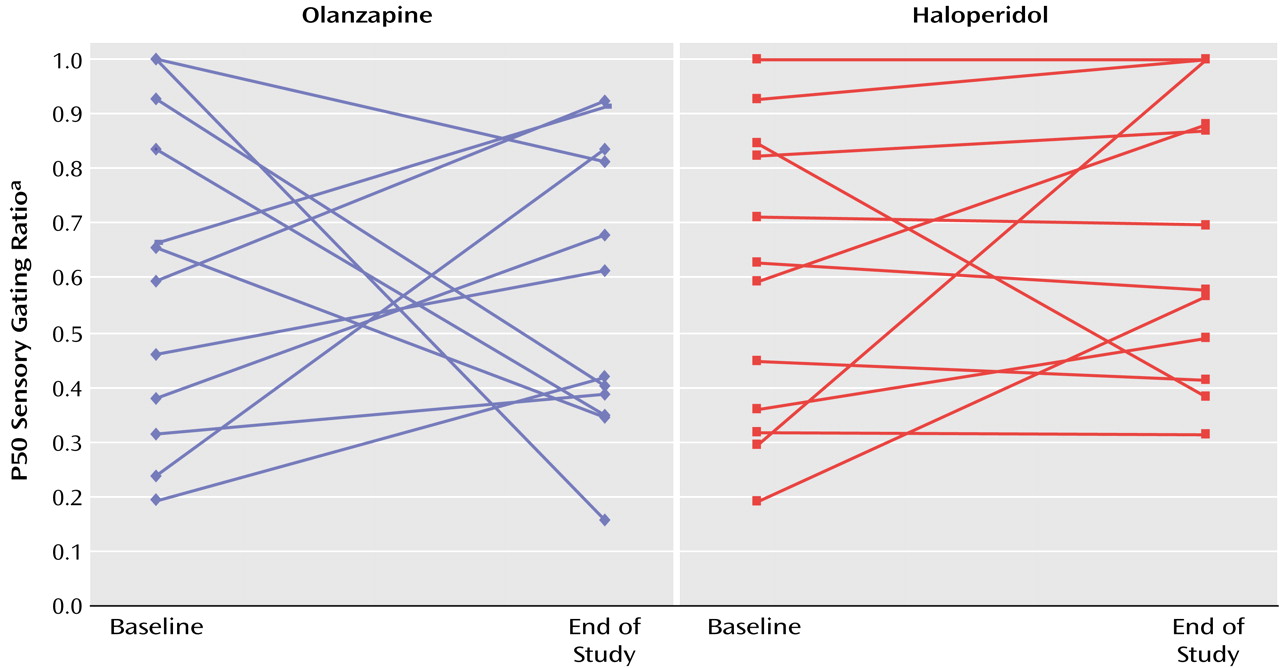
Twenty-seven patients entered the study. One patient dropped out before study completion, one was not capable of giving informed consent at the second P50 measurement occasion, and the baseline data of one patient were lost because of equipment malfunction. Data for 24 patients with complete measurements are presented. The haloperidol and olanzapine groups did not differ in terms of gender (10 male subjects in each group) or age (mean=46.1 [SD=10.0] and 41.5 years [SD=8.1], respectively). The mean haloperidol dose at the end of the study was 17.3 mg/day (SD=6.2); the mean olanzapine dose at the end of the study was 20.4 mg/day (SD=5.8).
The normal P50 sensory gating ratio cutoff in our laboratory is 0.50. The mean baseline ratios were above this cutoff for both the olanzapine and the haloperidol groups (mean=0.61 [SD=0.29] and 0.60 [SD=0.27], respectively). Seven patients in each group (58%) exceeded the normal sensory gating ratio cutoff. There were no between-group differences in P50 amplitudes or latencies following the first or second click at baseline.
The end-of-study P50 amplitude and latency measures did not significantly differentiate olanzapine-treated from haloperidol-treated patients nor did the end-of-study sensory gating ratios (mean=0.57 [SD=0.26] and 0.68 [SD=0.26], respectively) (
Figure 1). The time-by-drug interaction was not significant (F=0.68, df=1, 22, p=0.42). Six (50%) of the olanzapine-treated and four (33%) of the haloperidol-treated patients had normal sensory gating at endpoint. There was no significant group difference in the proportion of patients who had normal sensory gating at endpoint (χ
2=0.35, df=1, p=0.55). There were no significant within-group changes or between-group differences in BPRS total score, BPRS positive symptom score, or SANS total score (all p values >0.20).
Discussion
Olanzapine was not significantly superior to haloperidol for the treatment of sensory gating impairments. The medication-by-time effect for the P50 measure of sensory gating was not significant, nor was there a significant group difference in the number of subjects whose P50 suppression normalized with treatment. The failure to detect a beneficial effect of haloperidol on the P50 sensory gating index is consistent with previous research
(5).
The lack of a beneficial effect of olanzapine on the P50 sensory gating index differs from the findings of the Light et al. study
(7), which suggested that patients treated with new-generation antipsychotics, including olanzapine (N=6), but not conventional antipsychotics, have normal P50 suppression. The difference between the two studies may be related to the use of a randomized, longitudinal design in the current study versus cross-sectional assessment in the latter study. The lack of longitudinal assessment in the Light et al. study precludes attribution of group differences in P50 suppression to medication effects. The failure to detect a beneficial effect of olanzapine on P50 suppression is unlikely to be due to the relatively small number of subjects, since there was only a 6% reduction in sensory gating ratio in the olanzapine-treated patients, which even with a substantially larger number of subjects would not be statistically significant.
Our results are also in contradistinction to the putative effect of clozapine on P50 suppression. In previous longitudinal studies, clozapine has been shown to improve P50 suppression in a group of treatment-resistant patients
(5,
6). Improvement in sensory gating paralleled improvement in clinical status and decline in norepinephrine metabolite levels
(5,
6). The differential effect of clozapine and olanzapine on sensory gating may be related to the higher relative affinity of clozapine compared with olanzapine for the α-2 adrenergic receptor
(9). Alternatively, the lack of effect of olanzapine on P50 suppression could be related to the inclusion of treatment-resistant patients and the failure to observe a clinical response. However, there are several arguments against this explanation. First, there are a number of studies that have failed to show a relationship between P50 suppression and severity of negative or positive symptoms
(7,
10,
11). Second, the beneficial effect of clozapine on P50 suppression was observed in treatment-resistant patients. Conley and colleagues
(12) demonstrated that patients who fail to respond to olanzapine may respond to clozapine, which further argues for a differential effect of these drugs in this patient population.