A major advance in the treatment of schizophrenia and other psychoses was the introduction of long-acting depot formulations of antipsychotics in the 1960s. Depot agents have a number of advantages over oral medication
(1), including stabilization of serum drug levels, avoidance of first-pass metabolism, and assured medication delivery
(2,
3). These agents also allow well-controlled titration to the lowest effective doses. Maintaining consistent drug delivery with depot agents by reducing the variation between peak and trough plasma drug levels can improve patients’ responses to treatment, and lower peak plasma drug levels with depot than oral antipsychotics can reduce the incidence of adverse events
(2–
6).
Depot antipsychotics have been found to be generally well tolerated and more efficacious than their oral equivalents
(7,
8). Contrary to popular belief, some patients have reported a preference for depot antipsychotics over oral agents
(9). Compliance with a medication regimen has been found to improve when patients are switched to depot agents
(10), and these agents enable physicians to rapidly detect noncompliance.
Compliance, however, is not an all-or-nothing phenomenon. Patients with illnesses that require regular dosing of medications may vary in their compliance from taking all medications as prescribed to partial compliance or complete noncompliance
(1). Partial compliance is common among patients with schizophrenia
(11), as in other diseases requiring long-term treatment
(12,
13). Dolder et al.
(14) have recently shown that medication compliance (“adherence”) was better with atypical than typical antipsychotics in schizophrenia patients, but poor compliance was considerable even among those receiving the atypical antipsychotics. According to 12-month pharmacy refill records, 117 outpatients receiving haloperidol or perphenazine were without medication for approximately 7 days per month, while 171 patients receiving risperidone, olanzapine, or quetiapine were without medication for approximately 4 days per month. The difference in the compliance rate between atypical and typical antipsychotics was significant at 6 months (57% versus 50%, respectively; p=0.05) but not at 12 months (55% versus 50%; p=0.11).
Partial compliance is a serious problem that is often underappreciated by physicians. It is impossible to determine at what “threshold” of erratic medication taking an exacerbation will occur, and unprescribed or abrupt changes in dose can lead to unanticipated and unexplained adverse effects. For the patient with schizophrenia, the consequences of poor compliance can include demoralization, loss of confidence, loss of job, family discord, danger to self or others, and finally relapse and rehospitalization. The risk of relapse in first-episode patients has been shown to increase almost fivefold (risk ratio=4.89) when antipsychotic drug treatment was discontinued
(15). Thus, there is an urgent need to ensure continuous delivery of antipsychotic treatment for schizophrenia patients with a minimum of treatment interruptions.
Introduction of the atypical antipsychotics resulted in a substantial change in the paradigm for antipsychotic treatment. The superiority of the atypical agents over conventional neuroleptics in terms of both symptom change and tolerability is well documented
(16). Some have questioned whether outcome over time was affected by the introduction of the atypical agents. In a recent landmark study, however, rates of relapse during 1 year of treatment were significantly lower in patients with schizophrenia receiving risperidone than in patients receiving the most widely used conventional antipsychotic, haloperidol
(17). Atypical agents also have a better safety profile than conventional agents, with a greatly reduced risk of extrapyramidal symptoms and tardive dyskinesia
(18). However, as noted earlier
(14), poor compliance is common even with the atypical agents, and additional treatment options for patients with schizophrenia and other psychoses are needed. Specifically, many clinicians have expressed the need for a long-acting atypical antipsychotic
(19,
20).
Long-acting injectable risperidone is the first long-acting atypical agent to become available. It brings the advantages of long-acting formulations to those of an atypical agent. Pharmacokinetic studies have shown that fluctuations in plasma drug levels and peak plasma drug levels are lower with long-acting risperidone than after oral dosing
(21), indicating that the new formulation may provide more consistent and predictable plasma drug levels with superior tolerability.
The aim of the present placebo-controlled study was to evaluate the efficacy, safety, and tolerability of three different doses of long-acting injectable risperidone (25 mg, 50 mg, and 75 mg) in patients with schizophrenia.
Method
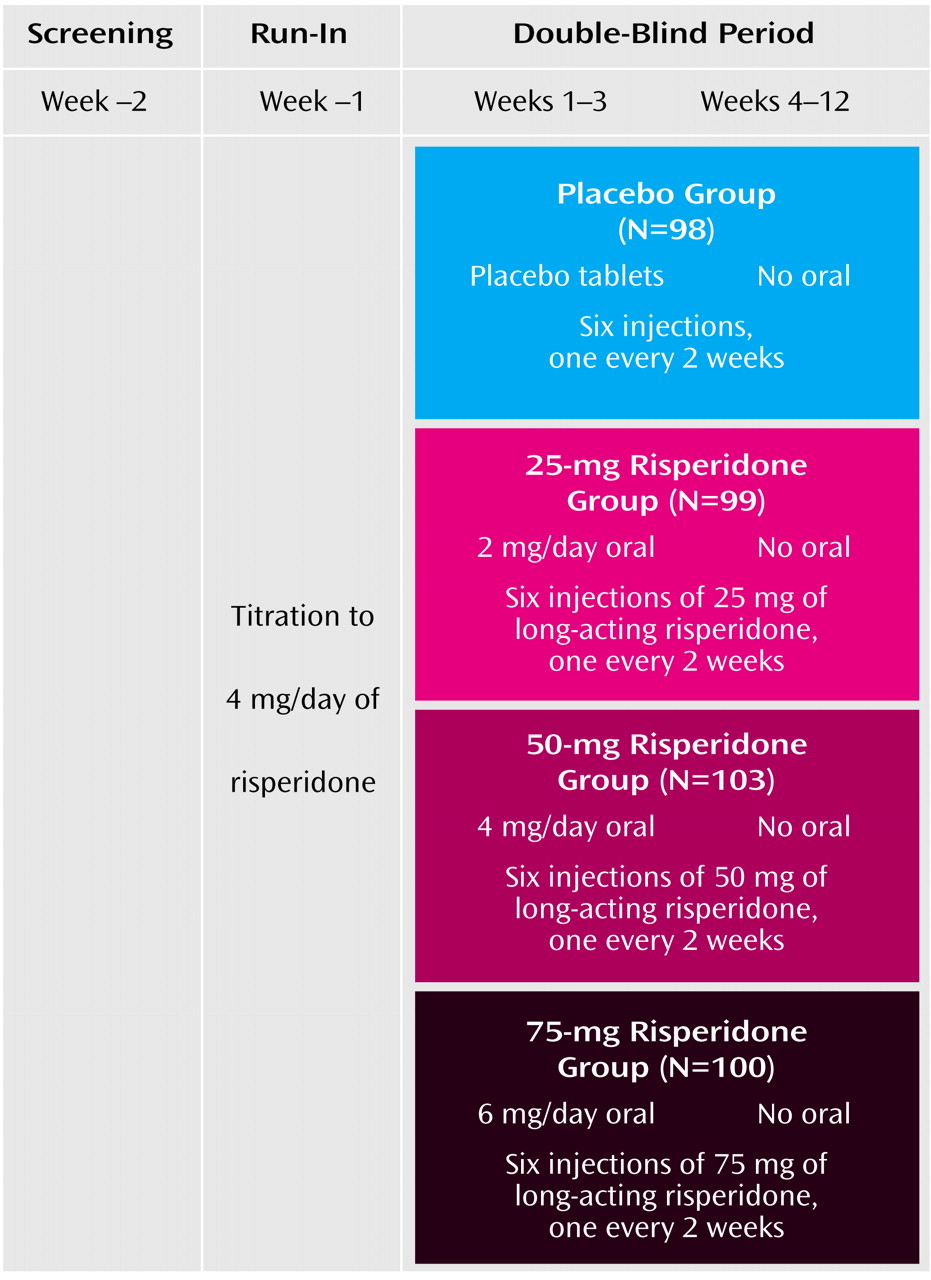
In a 12-week, multisite, randomized, double-blind, parallel-group study, patients with schizophrenia received intramuscular injections of long-acting risperidone (25 mg, 50 mg, or 75 mg) or placebo injections that were identical in appearance every 2 weeks. The trial design is summarized in
Figure 1.
After a 1-week screening period, doses of other oral antipsychotic medications were reduced and then discontinued. Simultaneously, oral risperidone was started at 2 mg/day and increased to 4 mg/day for at least 3 days. At the start of the 12-week double-blind phase, patients were randomly assigned to receive intramuscular injections of 25 mg, 50 mg, or 75 mg of long-acting risperidone or placebo every 2 weeks. Oral risperidone at doses shown in
Figure 1 or oral placebo was continued for the first 3 weeks of the double-blind phase. A dynamic method
(22) was used to randomly assign patients to treatment groups. Stratification factors included investigator, inpatient/outpatient status, and Positive and Negative Syndrome Scale
(23) total score at randomization.
The trial was conducted in accordance with current ICH–Good Clinical Practice guidelines and the Declaration of Helsinki and its subsequent revisions.
Trial Medication
Long-acting injectable risperidone is an aqueous suspension that contains risperidone in a matrix of glycolic acid–lactate copolymer. After intramuscular injection, the copolymer is gradually hydrolyzed at the injection site to ensure the slow, steady release of risperidone over a period of several weeks. The end-product of the copolymer hydrolysis is carbon dioxide and water. Single-dose studies of long-acting injectable risperidone show that plasma concentrations of the active moiety (risperidone plus 9-hydroxyrisperidone) start to increase gradually 3 weeks after injection, with peak levels being reached between weeks 4 and 6
(21). These pharmacokinetics imply that the optimal injection interval to produce the most stable plasma levels over time is 2 weeks. When starting treatment with long-acting injectable risperidone, initial antipsychotic coverage is required during the latency period after the first injection.
Subjects
Hospital outpatients or inpatients 18–55 years of age with a diagnosis of schizophrenia according to DSM-IV criteria were enrolled at 41 centers in the United States. Inclusion criteria included baseline Positive and Negative Syndrome Scale total scores of 60–120 and good general health, with standard laboratory test results within reference ranges or not clinically significant.
Patients were excluded from the trial if they had received a depot antipsychotic within 120 days of the start of the trial, were diagnosed as substance dependent, had tardive dyskinesia or a history of neuroleptic malignant syndrome, had a clinically significant ECG abnormality, were pregnant (or likely to become pregnant) or lactating, were at risk of violent behavior, or had current suicidal ideation. Patients who had a history of severe drug sensitivity or allergy, including sensitivity to risperidone, or who were unresponsive to risperidone were also excluded.
Written informed consent, consistent with local regulations, was obtained from each patient or a guardian or legal representative.
Measures of Efficacy and Safety
The primary measure of efficacy was the change from baseline (end of the run-in period) to endpoint (last-observation-carried-forward values) in total score on the Positive and Negative Syndrome Scale. All raters were trained in the use of the Positive and Negative Syndrome Scale, and interrater reliability was established before the start of the trial. Clinical improvement was defined a priori as a reduction of ≥20% in Positive and Negative Syndrome Scale total score. Positive and negative factor scores
(24) from the Positive and Negative Syndrome Scale are also reported. Positive and Negative Syndrome Scale evaluations were made at 2-week intervals. Each patient was rated weekly with the Clinical Global Impression (CGI) scale
(25).
Adverse events and vital signs were assessed at baseline and every 2 weeks. Serious adverse events were defined as those that resulted in death or were life-threatening, required hospitalization or prolongation of hospitalization, resulted in persistent or significant disability or incapacity, or resulted in a congenital anomaly or birth defect.
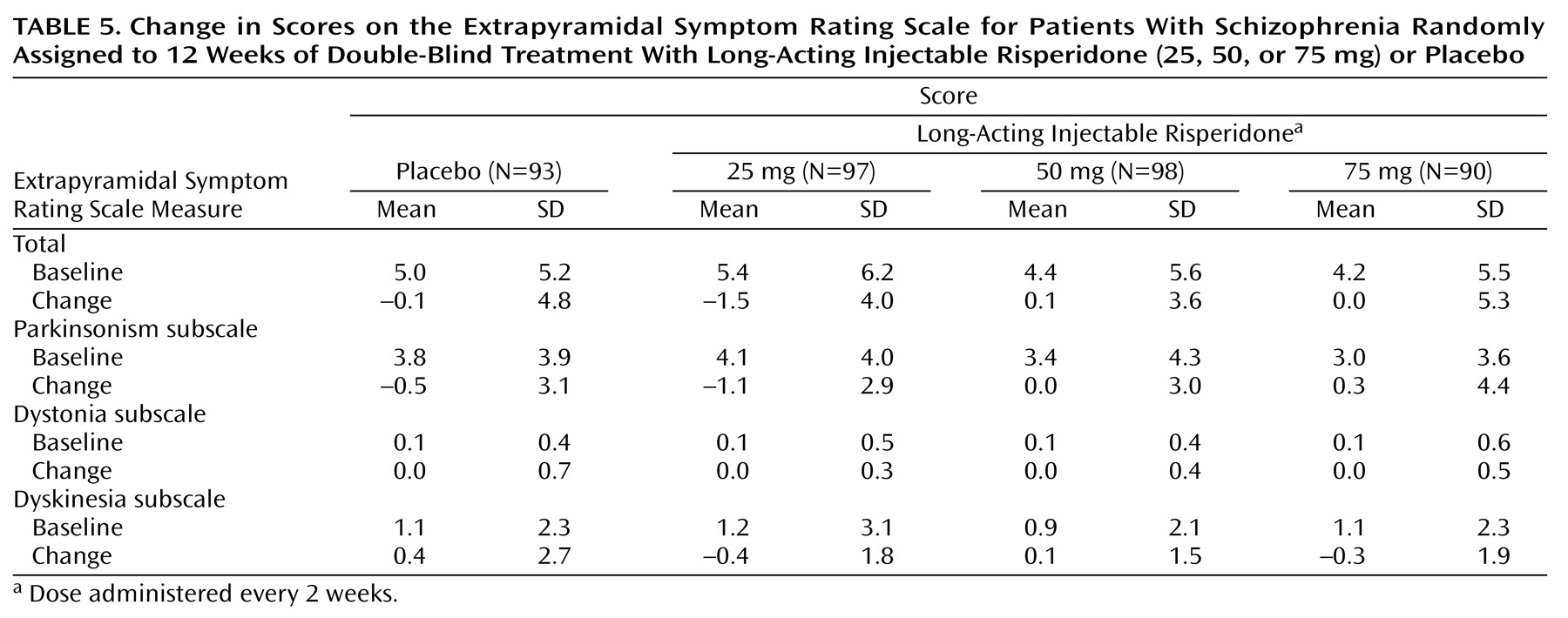
Spontaneously reported adverse events related to extrapyramidal symptoms were recorded during the trial. These include extrapyramidal disorder, hyperkinesia, hypertonia, tremor, hypokinesia, and involuntary muscle contractions. Severity of extrapyramidal symptoms was evaluated by means of the 55-item Extrapyramidal Symptom Rating Scale
(26). Investigators were trained in the use of the Extrapyramidal Symptom Rating Scale, and interrater reliability was established before the trial.
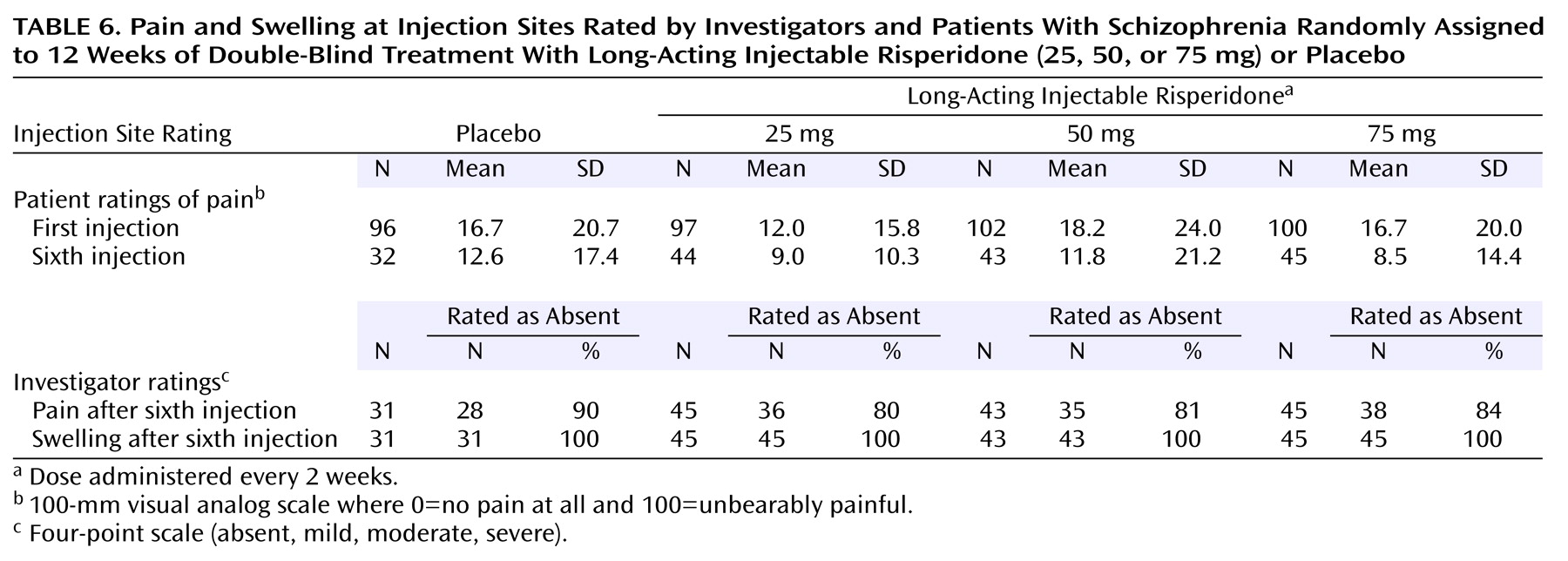
Patients evaluated pain at the injection site weekly and before and after each injection on a 100-mm visual analog scale (scored from 0 [no pain] to 100 [unbearably painful]). At the same time points, investigators rated injection-site pain, redness, swelling, and induration on a categorical scale (absent, mild, moderate, or severe).
Data Analysis
The primary efficacy population comprised patients who received at least one injection and had at least one postbaseline assessment during the double-blind phase. The primary safety population comprised all patients who received at least one injection during the double-blind phase.
For continuous efficacy measures, within-group changes were analyzed by paired t tests and between-group comparisons of change from baseline by an ANCOVA model
(27) including effects for treatment, investigator, and baseline value (or Positive and Negative Syndrome Scale stratification for analyses of CGI scores). Pairwise comparisons of each risperidone group with the placebo group were analyzed by using Dunnett’s test
(28). For categorical efficacy measures, between-group comparisons were analyzed by using Cochran-Mantel-Haenszel tests (with modified ridit scores)
(29) after we controlled for investigator and Positive and Negative Syndrome Scale stratification. Time to treatment discontinuation was assessed by Kaplan-Meier analysis
(30).
Results
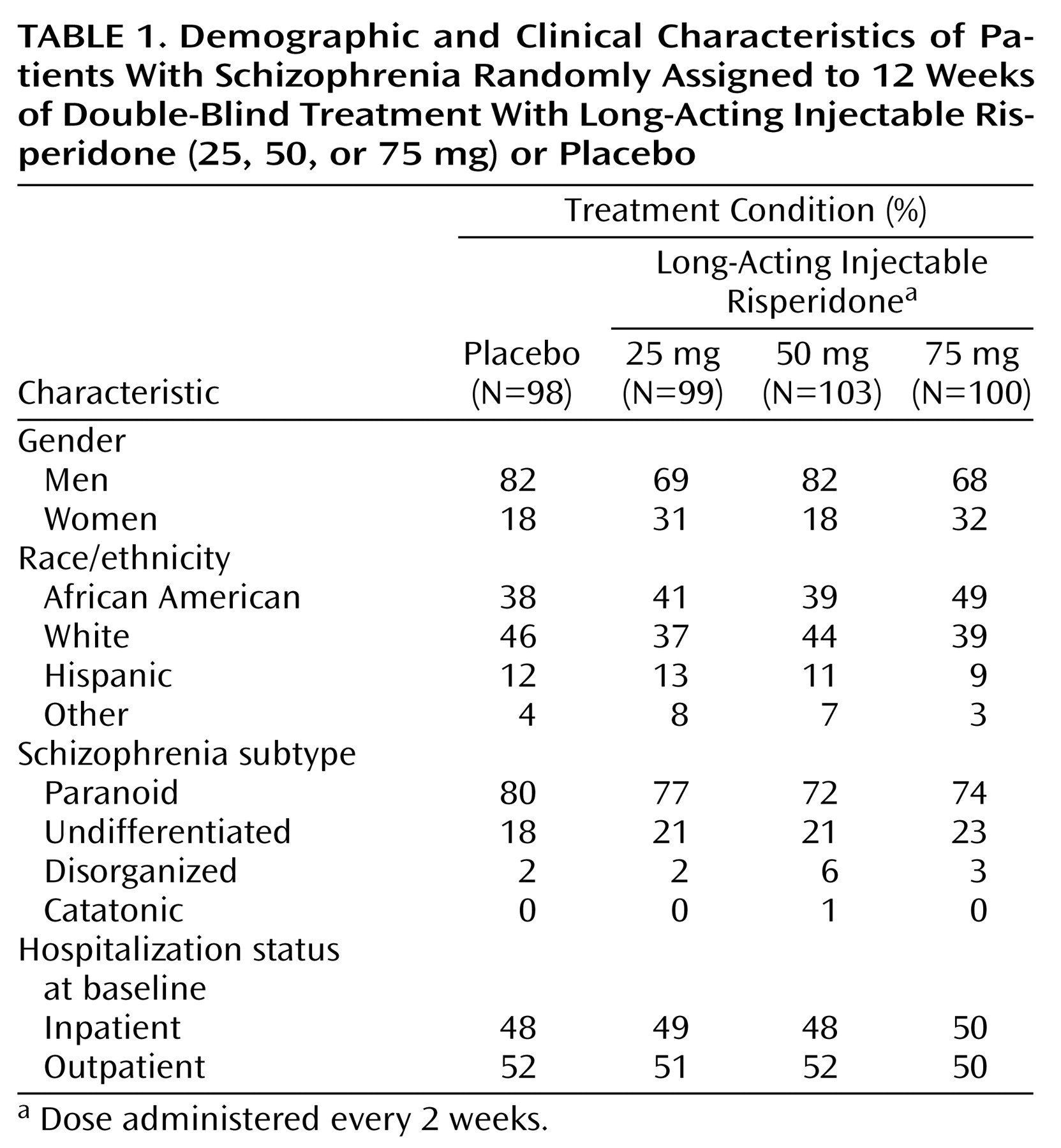
A total of 554 patients with schizophrenia were screened, of whom 461 entered the 1-week oral risperidone run-in period, and 400 initiated the double-blind phase. Of these 400 patients, 98 were randomly assigned to the placebo condition, and the remaining were assigned to the three doses of long-acting injectable risperidone (25 mg: N=99; 50 mg: N=103; 75 mg: N=100). The mean ages of the groups were 37.7 years (SD=9.4), 38.9 years (SD=9.8), 36.2 years (SD=9.5), and 38.1 years (SD=10.7), respectively. Efficacy was evaluated in the 370 patients with at least one postbaseline assessment. Background characteristics of the 400 patients who received at least one injection of long-acting risperidone are shown in
Table 1. Most of the patients (76%) had a diagnosis of paranoid schizophrenia. The number of previous hospitalizations was similar across the four groups (placebo [N=89]: median=4.0, range=0–28; risperidone, 25 mg [N=96]: median=3.5, range=0–99; risperidone, 50 mg [N=101]: median=4.0, range=0–50; risperidone, 75 mg [N=94]: median=4.0, range=0–63). Equal proportions were hospital outpatients and inpatients.
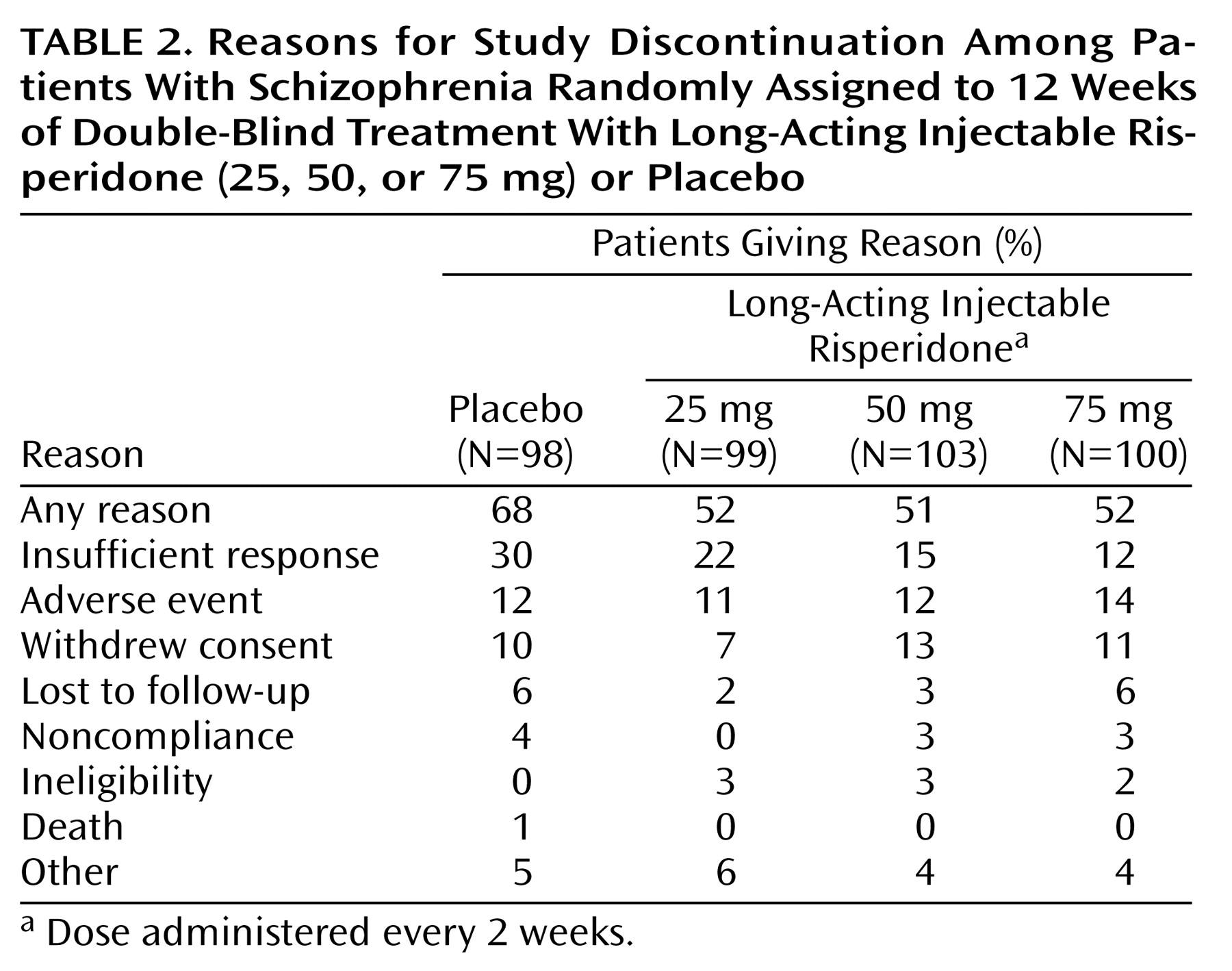
The trial was discontinued prematurely by 68% of the placebo patients and 51% to 52% of patients receiving long-acting risperidone. According to a Kaplan-Meier analysis, the dropout rate was similar in the four treatment groups during days 1–15, after which more placebo patients than patients receiving long-acting risperidone discontinued treatment. Reasons for discontinuation are listed in
Table 2.
Efficacy
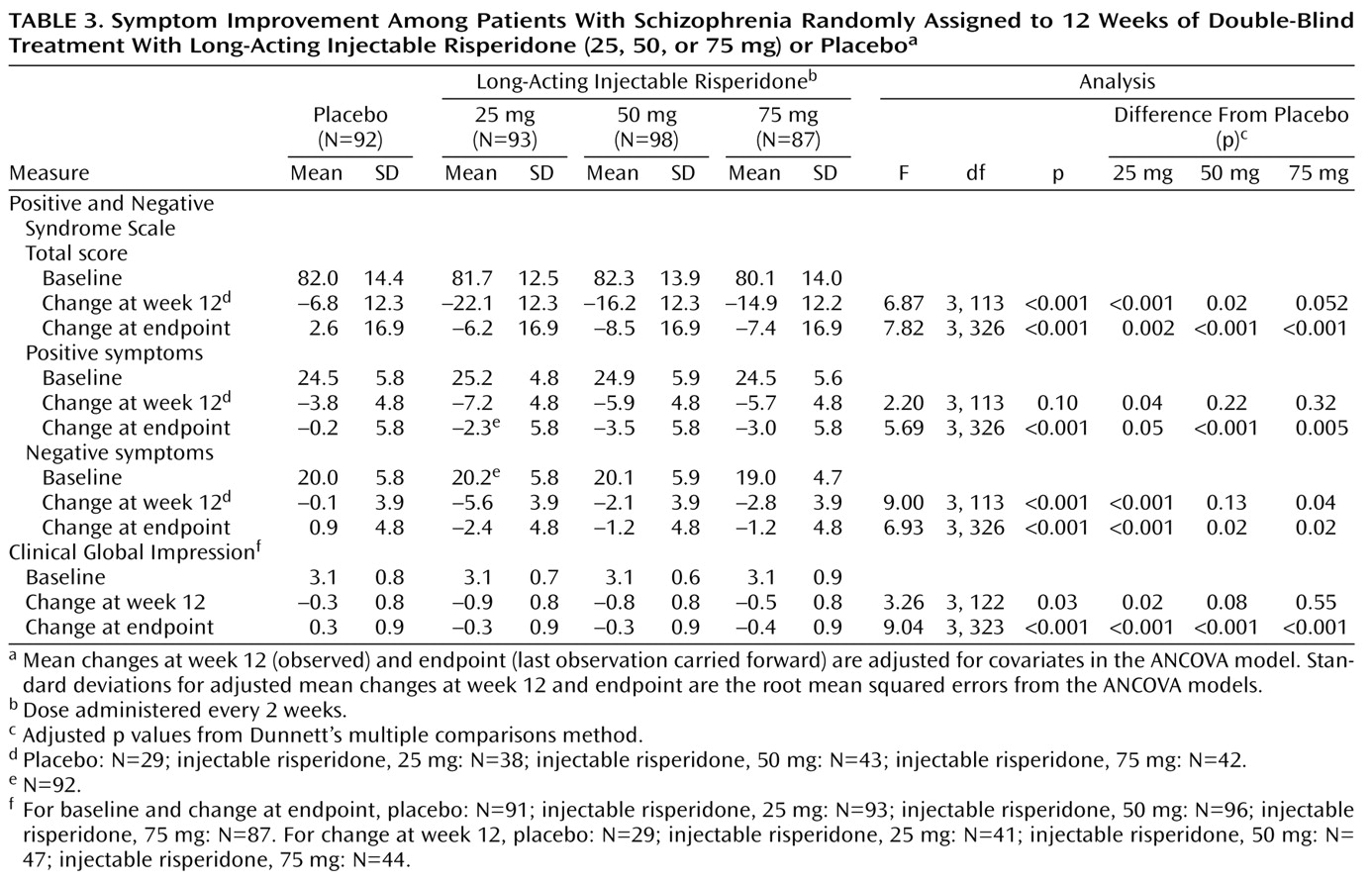
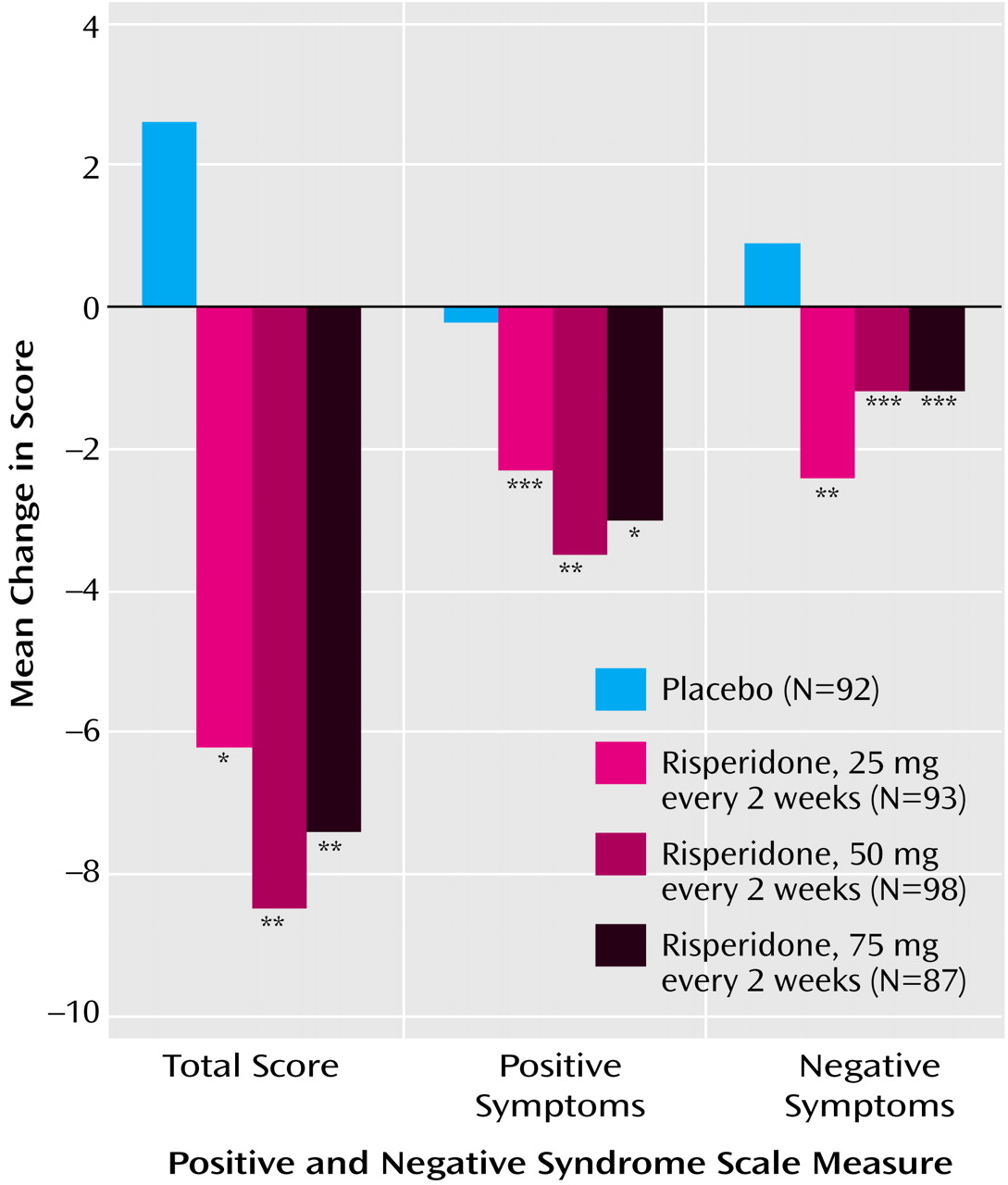
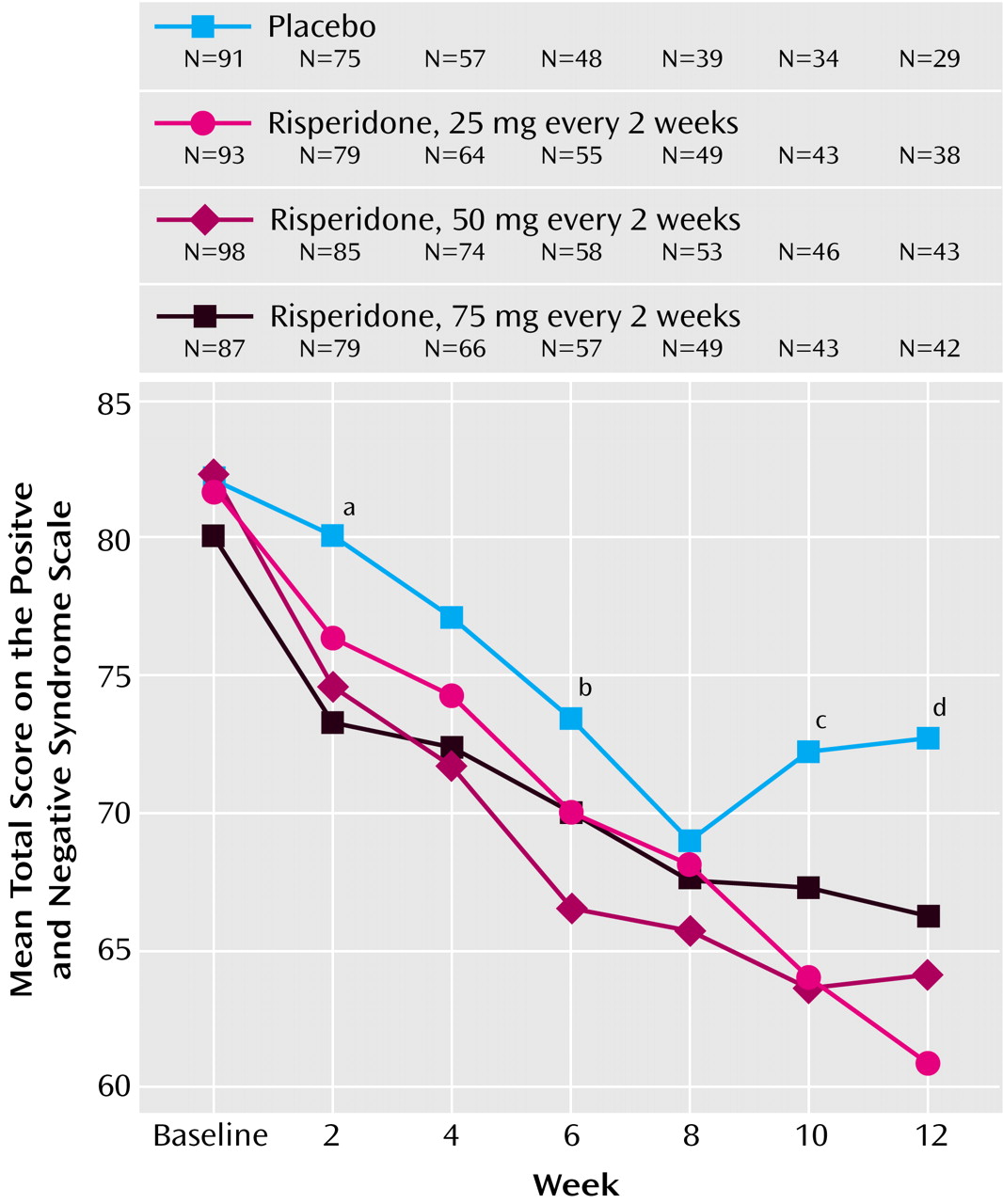
The mean total scores on the Positive and Negative Syndrome Scale at baseline ranged from 80.1 to 82.3 in the four treatment groups. Mean changes in Positive and Negative Syndrome Scale total scores from baseline to week 12 and endpoint, after adjusting for covariates in the ANCOVA model, are shown in
Table 3 and
Figure 2. At endpoint, comparison of each risperidone group versus placebo was significant (
Table 3). Improvements in both positive and negative symptoms were also substantial in the long-acting risperidone groups and significantly greater than in the placebo group (
Table 3,
Figure 2). Mean Positive and Negative Syndrome Scale total scores from baseline to week 12 (observed-case analysis) are shown in
Figure 3. Clinical improvement at endpoint (≥20% reduction in Positive and Negative Syndrome Scale total scores) was seen in 17% of placebo patients and 47%, 48%, and 39% of patients, respectively, in the three long-acting risperidone groups (12.10≤χ
2≤16.01, df=1, p<0.001). Significantly greater improvements in mean CGI severity scores from baseline to endpoint were shown by the three risperidone groups relative to that seen with placebo (
Table 3).
Safety and Tolerability
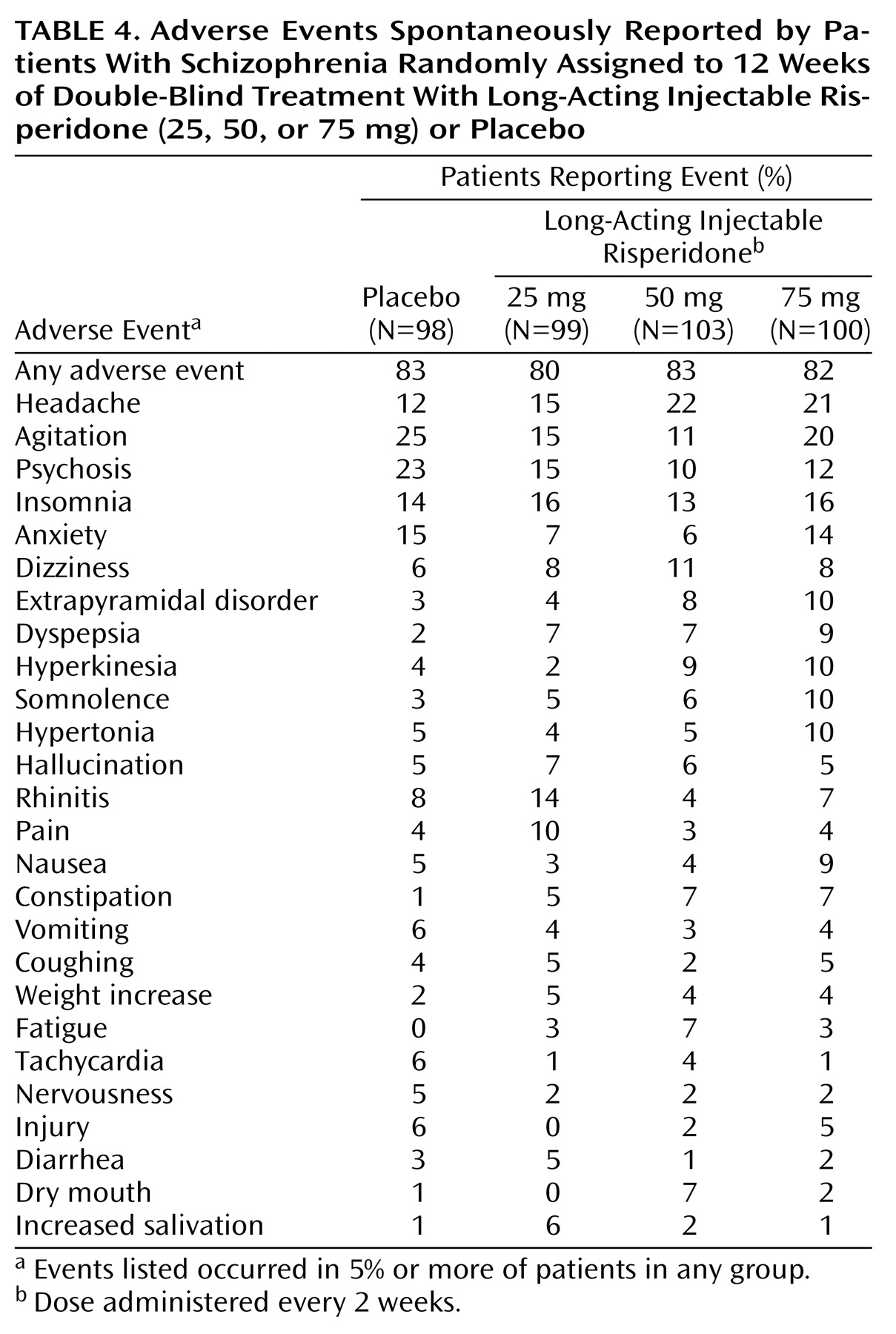
Similar proportions of patients in the placebo and long-acting risperidone groups (80% to 83%) reported adverse events. Adverse events reported by 5% or more of patients in any group are listed in
Table 4. Serious adverse events were more frequent in the placebo group (23.5%) than in the 25-mg, 50-mg, and 75-mg risperidone groups (13%, 14%, and 15%, respectively). In the placebo group, one death occurred as a result of injury.
The severity of extrapyramidal symptoms, as assessed by the Extrapyramidal Symptom Rating Scale, was mild at baseline and did not change during the trial (
Table 5). Adverse events related to extrapyramidal symptoms were spontaneously reported by 13% of patients in the placebo group, 10% in the 25-mg group, 24% in the 50-mg group, and 29% in the 75-mg group. Pairwise comparisons of each risperidone group with placebo were not significant (1.79≤χ
2≤2.59, df=1, p>0.10). During weeks 1–3 (when patients received oral and injectable medication), extrapyramidal symptoms were reported by 9% of the placebo group and 8%, 17%, and 18% of the 25-, 50-, and 75-mg risperidone groups, respectively. During weeks 4–12 (when patients received only injectable medication), the extrapyramidal symptoms rate was 9% in the placebo group and 3%, 14%, and 23% in the 25-, 50-, and 75-mg groups.
Changes in body weight at endpoint were small and appeared to be dose related (0.5 kg, 1.2 kg, and 1.9 kg in the 25-mg, 50-mg, and 75-mg groups, respectively, and –1.4 kg in the placebo group) (F=9.24, df=3, 297, p<0.001). Differences between placebo and any risperidone group with regard to QT interval or other measures of cardiovascular safety were not significant. Discontinuations as a result of adverse events were similar in patients receiving long-acting risperidone and placebo (
Table 2).
Patients’ perception of injection-site pain was low at the beginning of the trial and decreased even further during long-acting risperidone treatment (
Table 6). According to investigator assessments, most patients had no pain and no swelling after the sixth injection (
Table 6).
Concomitant Medications
Similar proportions of patients receiving placebo (82%) and long-acting injectable risperidone (86%) used concomitant medication during the double-blind phase. Antiparkinsonian medication was received by 13% of placebo patients, 12% of patients in the 25-mg risperidone group, and by 23% of patients in both the 50-mg and 75-mg groups. Use of antiparkinsonian medication was lower during weeks 1–12 of the double-blind phase (18% of patients) than during the 1-week oral run-in period (24%). Concomitant antipsychotics other than risperidone were not permitted during the trial, although restricted use of sedative medications (including lorazepam, chloral hydrate, and temazepam) was allowed. Fewer patients in the 25-mg (43%) and 50-mg (45%) groups than patients in the placebo (51%) or 75-mg (57%) groups received sedatives. Twelve percent of the placebo patients and 15%, 18%, and 20% of patients in the 25-mg, 50-mg, and 75-mg groups, respectively, received antidepressants. Use of β-blocking agents was similar in the placebo (3%), 25-mg (5%), and 50-mg (3%) groups and was higher in the 75-mg group (10%).
Discussion
In this 12-week study of patients with schizophrenia (both hospital outpatients and inpatients), long-acting intramuscular risperidone (25 mg, 50 mg, or 75 mg every 2 weeks) was significantly more efficacious than placebo in reducing the signs and symptoms of schizophrenia, as assessed by the change in scores on the Positive and Negative Syndrome Scale measures and the CGI.
Long-acting injectable risperidone was well tolerated, with minimal weight change and a safe cardiovascular profile. Severity of extrapyramidal symptoms (Extrapyramidal Symptom Rating Scale score) was low at baseline with minimum change between baseline and endpoint. The incidence of spontaneously reported adverse events related to extrapyramidal symptoms was comparable in the placebo (13%) and the 25-mg risperidone (10%) groups, with higher rates in the 50-mg (24%) and 75-mg (29%) groups. This lack of difference in extrapyramidal symptoms rates between placebo and the 25-mg dose was not confounded by an increased use of antiparkinsonian medication: antiparkinsonian medications were received by similar proportions of placebo (13%) and 25-mg risperidone patients (12%) and by 23% of both the 50-mg and 75-mg groups.
According to patient ratings, injection site pain was generally mild throughout the trial and was diminished from the first injection to the last in all four treatment groups. Thus, pain at the injection site is unlikely to be a factor in determining patients’ acceptance of long-acting injectable risperidone. This represents an important advantage over conventional oil-based depot formulations, which are often associated with pain and other adverse events at the injection site (31, 32).
To maintain steady plasma levels of the active moiety throughout the randomized period, patients in the three long-acting risperidone groups received 2, 4, or 6 mg/day of oral risperidone during the latency period after the first injection before the main release of risperidone began. This meant that for patients in the 25-mg arm, the dose of 4 mg of oral risperidone during the run-in period was reduced to 2 mg for the 3 weeks after the first injection. This might explain the similar dropout rates in the placebo and 25-mg groups during the first 2 weeks after randomization (
Figure 2).
The study results indicate that 25 mg of long-acting injectable risperidone given every 2 weeks appears to offer the optimum risk/benefit profile for most patients requiring maintenance treatment with an antipsychotic. The 25-mg dose was well tolerated (for example, the extrapyramidal symptoms rate was similar in the placebo and 25-mg groups) and efficacious, particularly with regard to negative symptoms. Over 45% of patients in the 25-mg risperidone group showed a ≥20% improvement in Positive and Negative Syndrome Scale total scores. The 75-mg dose, although efficacious, offered no incremental benefit over the 25-mg and 50-mg doses.
Long-acting injectable risperidone appears to combine the most valuable features of an atypical antipsychotic (broadly efficacious and well tolerated) with those of injectable long-acting antipsychotics (improved bioavailability and assured medication delivery). Long-acting injectable risperidone provides both clinicians and patients (and their families) with a new mode of maintenance treatment that can improve the outcome of long-term therapy for patients with schizophrenia and other chronic psychoses.
Acknowledgments
The principal investigators of the Ris-USA-121 trial were as follows: Adityanjee, M.D., Cleveland; Scott Balogh, M.D., Augusta, Ga.; Nigel Bark, M.D., Bronx, N.Y.; George Bartzokis, M.D., North Little Rock, Ark.; Bijan Bastani, M.D., Beachwood, Ohio; Sally Berry, M.D., Atlanta; Ronald Brenner, M.D., Cedarhurst, N.Y.; David Brown, Austin, Tex.; Matthew Byerly, M.D., Dallas; Jose Canive, M.D., Albuquerque, N.Mex.; Amal Chakraburtty, M.D., Oklahoma City; George Chappell, M.D., Olympia, Wash.; David Daniel, M.D., Falls Church, Va.; Eduardo Dunayevich, M.D., Cincinnati; Louis Fabre, M.D., Houston; David Feifel, M.D., San Diego; Rohan Ganguli, M.D., Pittsburgh; Ira Glick, M.D., Stanford, Calif.; Jeffrey Grace, M.D., Buffalo; Raquel Gur, M.D., Philadelphia; Uriel Halbreich, M.D., Buffalo; Mark Hamner, M.D., Charleston, S.C.; Carolyn Heimberg, M.D., Philadelphia; Philip Janicak, M.D., Chicago; Steven Lamberti, M.D., Rochester, N.Y.; Ronald Landbloom, M.D., St. Paul; Mark Lerman, M.D., Naperville, Ill.; Michael Lesem, M.D., Houston; Jean-Pierre Lindenmayer, M.D., New York; Adam Lowy, M.D., Washington, D.C.; Joseph McEvoy, M.D., Butner, N.C.; Denis Mee-Lee, M.D., Honolulu; Herbert Meltzer, M.D., Nashville, Tenn.; Alexander Miller, M.D., San Antonio, Tex.; Henry Nasrallah, M.D., Jackson, Miss.; Jorg Pahl, M.D., Oklahoma City; Anand Pandurangi, M.D., Richmond, Va.; Michael Plopper, M.D., San Diego, Calif.; Sheldon Preskorn, M.D., Wichita, Kan.; Jeffrey Rausch, M.D., Augusta, Ga.; Ralph Richter, M.D., Tulsa, Okla.; David Sack, M.D., Cerritos, Calif.; Alan Schneider, M.D., Los Angeles; Michael Smith, M.D., Torrance, Calif.; Kenneth Sokolski, M.D., Anaheim, Calif.; Marshall Thomas, M.D., Denver; Seetharaman Vivek, M.D., Jamaica, N.Y.