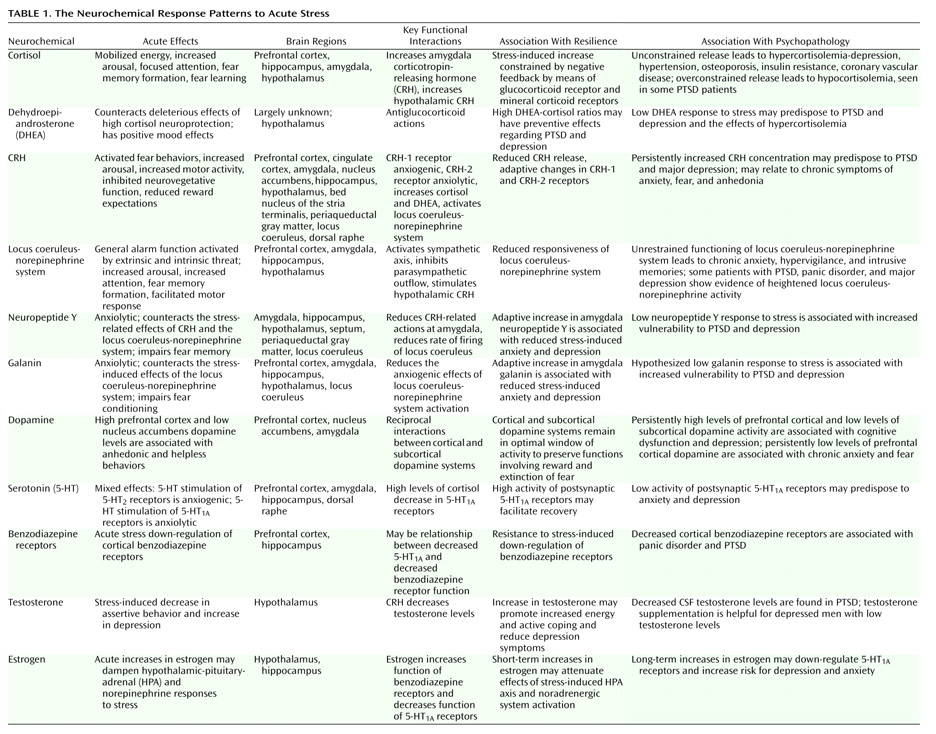
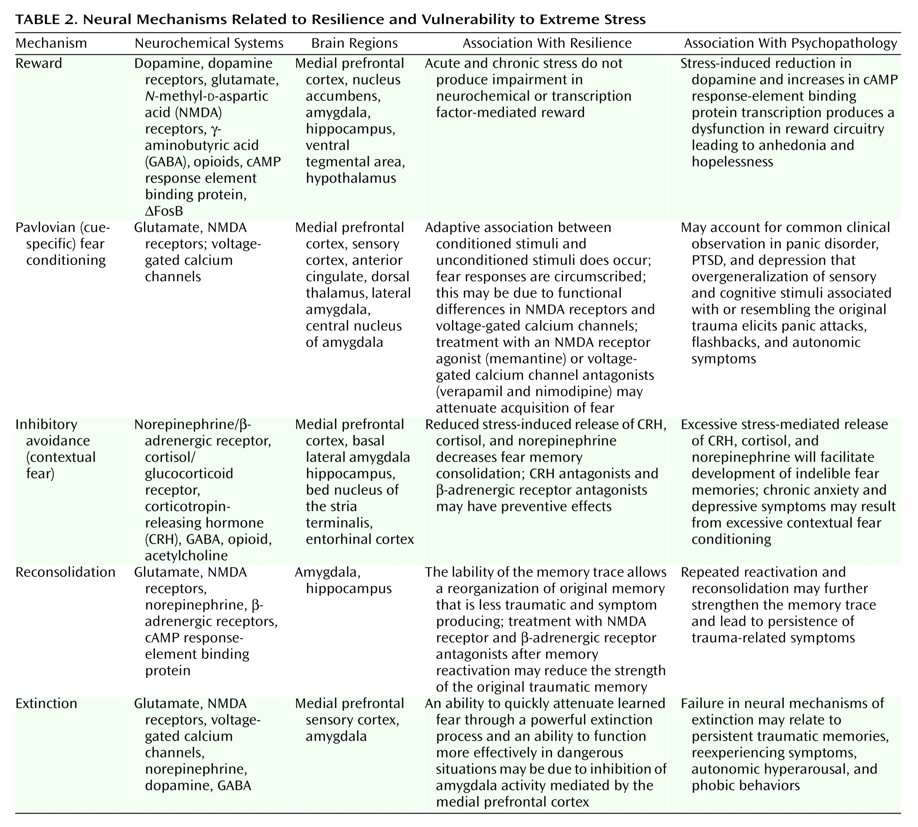
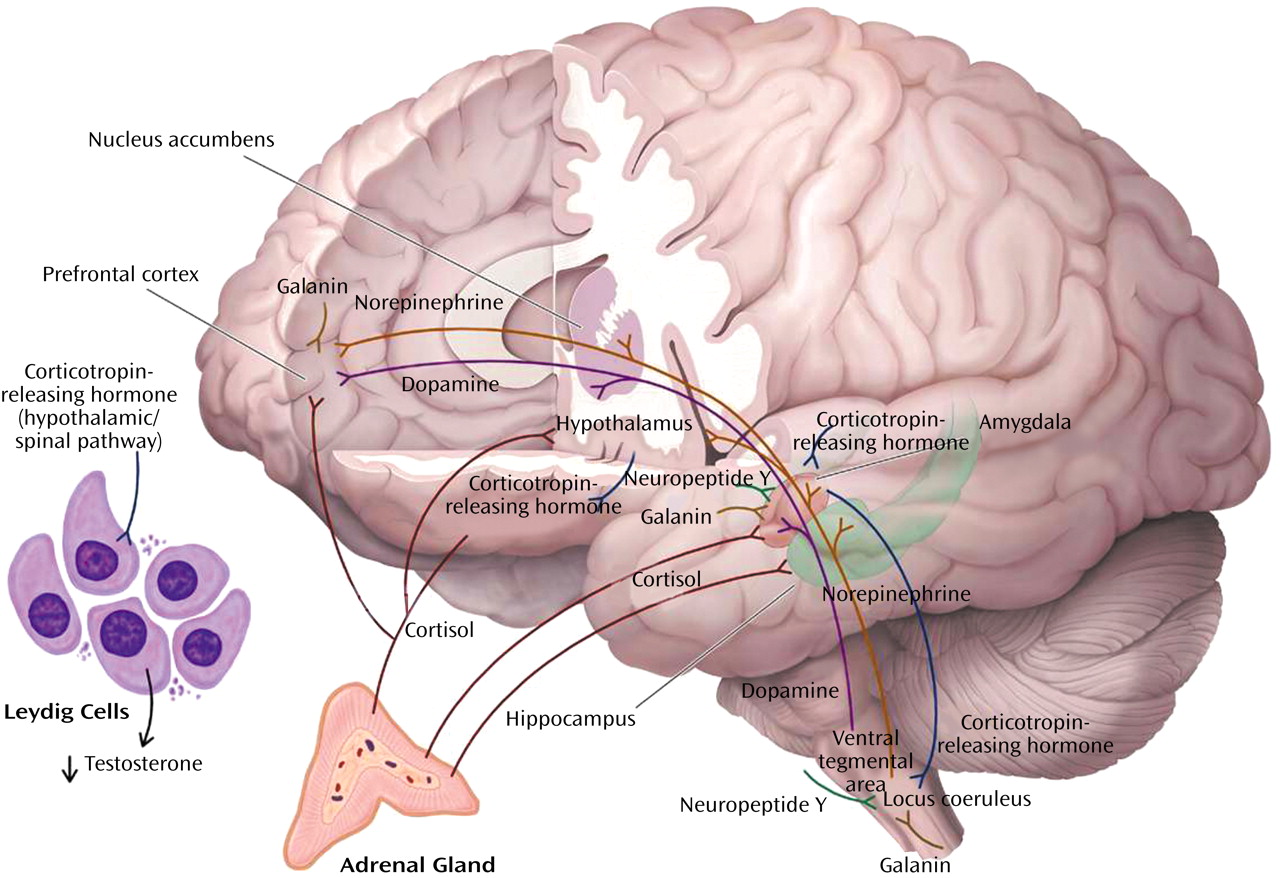
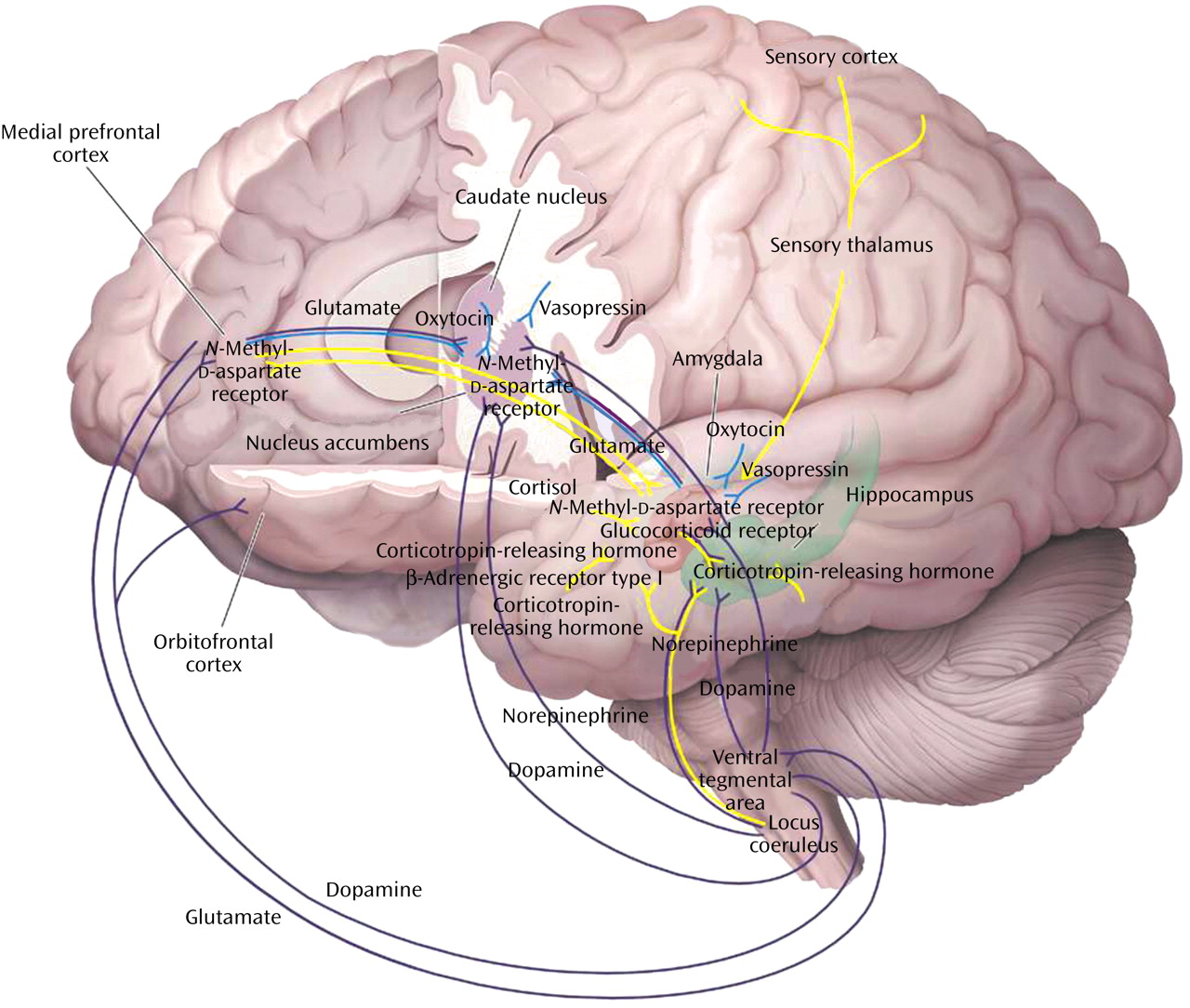
Fear Conditioning
Fear conditioning in many patients with PTSD and major depression causes vivid recall of memories of traumatic events, autonomic hyperarousal, and even flashbacks elicited by sensory and cognitive stimuli associated with prior traumas. Consequently, patients may begin to avoid these stimuli in their everyday lives, or a numbing of general emotional responsiveness may ensue. Resilience to the effects of severe stress may be characterized by the capacity to avoid overgeneralizing specific conditioned stimuli to a larger context, reversible storage of emotional memories, and facilitated extinction.
Classical fear conditioning is a form of associative learning in which subjects come to express fear responses to a neutral conditioned stimulus that is paired with an aversive unconditioned stimulus. The conditioned stimulus, as a consequence of this pairing, acquires the ability to elicit a spectrum of behavioral, autonomic, and endocrine responses that normally would only occur in the context of danger
(156). Fear conditioning can be adaptive and enable efficient behavior in dangerous situations. The individual who can accurately predict threat can engage in appropriate behaviors in the face of danger. In the clinical situation, specific environmental features (conditioned stimuli) may be linked to the traumatic event (unconditioned stimuli), such that reexposure to a similar environment produces a recurrence of the symptoms of anxiety and fear. Patients often generalize these cues and experience a continuous perception of threat to the point that they become conditioned to context.
Cue-specific conditioned stimuli are transmitted to the thalamus by external and visceral pathways. Afferents then reach the lateral amygdala by means of two parallel circuits: a rapid subcortical path directly from the dorsal (sensory) thalamus and a slower regulatory cortical pathway encompassing the primary somatosensory cortices, the insula, and the anterior cingulate/prefrontal cortex. Contextual conditioned stimuli are projected to the lateral amygdala from the hippocampus and perhaps the bed nucleus of the stria terminalis. The long loop pathway indicates that sensory information relayed to the amygdala undergoes substantial higher-level processing, thereby enabling assignment of significance based on prior experience to complex stimuli. Cortical involvement in fear conditioning is clinically relevant because it provides a mechanism by which cognitive factors will influence whether symptoms are experienced or not following exposure to stress
(157).
During the expression of fear-related behaviors, the lateral amygdala engages the central nucleus of the amygdala, which, as the principal output nucleus, projects to areas of the hypothalamus and brainstem that mediate the autonomic, endocrine, and behavioral responses associated with fear
(158). The molecular and cellular mechanisms that underlie synaptic plasticity in amygdala-dependent learned fear are an area of active investigation
(159). Long-term potentiation in the lateral amygdala appears to be a critical mechanism for storing memories of the association between conditioned stimuli and unconditioned stimuli
(156). A variety of behavioral and electrophysiological data have led LeDoux and colleagues
(157,
158) to propose a model to explain how neural responses to the conditioned stimuli and unconditioned stimuli in the lateral amygdala could influence long-term potentiation-like changes that store memories during fear conditioning. This model proposes that calcium entry through NMDA receptors and voltage-gated calcium channels initiates the molecular processes to consolidate synaptic changes into long-term memory
(156). Short-term memory requires calcium entry only through NMDA receptors and not voltage-gated calcium channels.
This hypothesis leads to several predictions that may have relevance to psychological responses to stress. It suggests that blocking NMDA receptors in the amygdala during learning should impair memory of short- and long-term fear. This has been demonstrated in rodents
(160,
161). Valid human models of fear conditioning and the availability of the NMDA receptor antagonist memantine should permit this hypothesis to be tested clinically
(162). If memantine impairs the acquisition of fear in humans, it may have use in the prevention and treatment of stress-induced disorders such as PTSD. Blockade of voltage-gated calcium channels appears to block long-term but not short-term memory
(163). Therefore, clinically available calcium channel blockers such as verapamil and nimodipine may be helpful in diminishing the intensity and impact of recently acquired fear memory and perhaps in preventing PTSD as well.
This discussion has focused primarily upon the neural mechanisms related to the coincident learning of the unconditioned stimuli-conditioned stimuli association (i.e., Pavlovian fear conditioning) in the lateral amygdala. However, there is significant evidence that a broader neural circuitry underlies the memory of fear that is modulated by amygdala activity. The inhibitory-avoidance paradigm is used to examine memory consolidation for aversively motivated tasks and involves intentional instrumental choice behavior. Studies using inhibitory avoidance learning procedures have been used to support the view that the amygdala is not the sole site for fear learning but, in addition, can modulate the strength of memory storage in other brain structures
(164).
There is evidence that Pavlovian fear conditioning and inhibitory avoidance involve fundamentally different neural mechanisms. Pavlovian fear conditioning and inhibitory avoidance are differentially affected by posttraining pharmacological manipulations. The two types of learning involve different experimental procedures. In Pavlovian fear conditioning, the presentation of the conditioned stimuli and unconditioned stimuli occurs independent of behavior, whereas with inhibitory avoidance shock, delivery is contingent upon an animal’s behavioral response. Inhibitory avoidance may involve a more complex neural network because an animal’s response is contingent upon a number of contextual cues, in contrast to the more specific conditioned stimuli and unconditioned stimuli. The basal lateral amygdala is the primary amygdala nucleus responsible for voluntary emotional behavior based upon aversive emotional events, whereas the central nucleus of the amygdala is more involved in Pavlovian responses to fear-inducing stimuli
(165). The relevance of the inhibitory-avoidance paradigm to human fear and anxiety rests on its assessment of a behavioral response to a fear-inducing context
(166).
Specific drugs and neurotransmitters infused into the basal lateral amygdala influence consolidation of memory for inhibitory avoidance training. Posttraining peripheral or intra-amygdala infusions of drugs affecting GABA, opioid, glucocorticoid, and muscarinic acetylcholine receptors have dose- and time-dependent effects on memory consolidation
(164). Norepinephrine infused directly into the basal lateral amygdala after inhibitory avoidance training enhances memory consolidation, indicating that the degree of activation of the noradrenergic system within the amygdala by an aversive experience may predict the extent of the long-term memory for the experience
(167).
Interactions among CRH, cortisol, and norepinephrine receptors have important effects on memory consolidation, which is likely to be relevant to the effects of traumatic stress on memory. Extensive evidence indicates that glucocorticoids influence long-term memory consolidation by means of stimulation of glucocorticoid receptors. The glucocorticoid effects on memory consolidation require activation of the basal lateral amygdala, and lesions of the basal lateral amygdala block retention enhancement of intrahippocampal infusions of a glucocorticoid receptor agonist. Additionally, the basal lateral amygdala is a critical locus of interaction between glucocorticoids and norepinephrine in modulating memory consolidation
(168).
There is also extensive evidence consistent with a role for CRH in mediating the effects of stress on memory consolidation. Activation of CRH receptors in the basal lateral amygdala by CRH released from the central nucleus of the amygdala facilitates the effects of stress on memory consolidation. As reviewed, there are important functional interactions between the CRH and norepinephrine systems, including a role in memory consolidation. Memory enhancement produced by CRH infusions in the hippocampus are blocked by propranolol and the noradrenergic toxin DSP-4 (75-R), suggesting that CRH infusions by means of a presynaptic mechanism stimulate norepinephrine release in the hippocampus
(169).
Efferent projections from the basal lateral amygdala are also crucial to memory formation. The basal lateral pathway of the amygdala stria terminalis is involved, since lesions of the stria terminalis impair the memory-enhancing effects of intra-amygdala infusions of norepinephrine and systemic dexamethasone, which are presumably acting on the hippocampus. Also, lesions of the nucleus accumbens block the memory-enhancing effects of intra-amygdala infusions of glucocorticoid receptor agonist. Finally, the cortex is also a locus for memory consolidation, since projections from the basal lateral amygdala are essential in the modulation of memory by the entorhinal cortex
(165,
170).
These results support the concept that CRH, by means of an interaction with glucocorticoids, interacts with the noradrenergic system to consolidate traumatic memories. Individuals with excessive stress-induced release of CRH, cortisol, and norepinephrine are likely to be prone to the development of indelible traumatic memories and their associated reexperiencing symptoms. Administration of CRH antagonists, glucocorticoid receptor antagonists, and β-adrenergic receptor antagonists may prevent these effects in vulnerable subjects.
Reconsolidation
Reconsolidation is a process in which old, reactivated memories undergo another round of consolidation
(171–
173). The process of reconsolidation is extremely relevant to both vulnerability and resiliency to the effects of extreme stress. It is the rule rather than the exception that memories are reactivated by cues associated with the original trauma. Repeated reactivation of these memories may serve to strengthen the memories and facilitate long-term consolidation
(174,
175). Each time a traumatic memory is retrieved, it is integrated into an ongoing perceptual and emotional experience and becomes part of a new memory. Moreover, preclinical studies indicate that consolidated memories for auditory fear conditioning, which are stored in the amygdala
(176), hippocampal-dependent contextual fear memory
(171), and hippocampal-dependent memory associated with inhibitory avoidance
(172) are sensitive to disruption upon reactivation by administration of a protein synthesis inhibitor directly into the amygdala and hippocampus, respectively. The reconsolidation process, which has enormous clinical implications, results in reactivated memory trace then returns to a state of lability and must undergo consolidation once more if it is to remain in long-term storage. Some controversies persist regarding the temporal persistence of systems reconsolidation. Debiec and colleagues
(171) found that intrahippocampal infusions of anisomycin caused amnesia for a consolidated hippocampal-dependent memory if the memory was reactivated, even up to 45 days after training. Milekic and Alberini
(172) however, found that the ability of intrahippocampal infusion of anisomycin to produce amnesia for an inhibitory avoidance task was evident only when the memory was recent (up to 7 days old). Further work is needed to resolve this very important question
(173).
The reconsolidation process involves NMDA receptors and β-adrenergic receptors and requires cAMP response-element binding protein induction. The cAMP response- element binding protein requirement suggests that nuclear protein synthesis is necessary
(177). NMDA receptor antagonists and β receptor antagonists impair reconsolidation
(174,
178). The effect of the β receptor antagonist propranolol was greater after memory reactivation than when administered immediately after the initial training. These results suggest that reactivation of memory initiates a cascade of intracellular events that involve both NMDA receptor and β receptor activation in a fashion similar to postacquisition consolidation.
This remarkable lability of a memory trace, which permits a reorganization of an existing memory in a retrieval environment, provides a theoretical basis for both psychotherapeutic and pharmacotherapeutic intervention for traumatic stress exposure. Administration of β receptor and NMDA receptor antagonists shortly after the initial trauma exposure as well as after reactivation of memory associated with the event may reduce the strength of the original traumatic memory.
Extinction
When the conditioned stimuli are presented repeatedly in the absence of the unconditioned stimuli, a reduction in the conditioned fear response occurs. This process is called extinction. It forms the basis for exposure-based psychotherapies for the treatment of a variety of clinical conditions characterized by exaggerated fear responses. Individuals who show an ability to attenuate learned fear quickly through powerful and efficient extinction processes are likely to function more effectively under dangerous conditions. They may also be less susceptible to the effects of intermittent exposure to fear stimuli, which can reinstate fear-conditioned learning. Highly stress-resilient individuals under extreme stress generally experience fear but have the capacity to function well under states of high fear. In addition, individuals in positions that regularly cause them to confront danger need to be able to extinguish learned fears rapidly.
Extinction is characterized by many of the same neural mechanisms as in fear acquisition. Activation of amygdala NMDA receptors by glutamate is essential
(179), and L-type voltage-gated calcium channels also contribute to extinction plasticity
(180). Long-term extinction memory is altered by a number of different neurotransmitter systems, including GABA, norepinephrine, and dopamine, in a manner similar to fear acquisition
(181,
182).
Destruction of the medial prefrontal cortex blocks recall of fear extinction
(183,
184), indicating that the medial prefrontal cortex might store long-term extinction memory. Infralimbic neurons, which are part of the medial prefrontal cortex, fire only when rats are recalling extinction—greater firing correlates with reduced fear behaviors
(185). It has been suggested that the consolidation of extinction involves potentiation of inputs into the medial prefrontal cortex by means of NMDA-dependent plasticity. The basal lateral amygdala sends direct excitatory inputs to the medial prefrontal cortex, and NMDA antagonists infused into the basal lateral amygdala block extinction. The ability of the medial prefrontal cortex to modulate fear behaviors is probably related to projections from the medial prefrontal cortex by means of GABA interneurons to the basal lateral amygdala
(186).
Failure to achieve an adequate level of activation of the medial prefrontal cortex after extinction might lead to persistent fear responses
(187). Individuals with the capacity to function well after experiencing states of high fear may have potent medial prefrontal cortex inhibition of amygdala responsiveness. In contrast, patients with PTSD exhibit depressed ventral medial prefrontal cortex activity, which correlates with increased autonomic arousal after exposure to traumatic reminders (unpublished work by Bremner et al.). Consistent with this hypothesis, we
(188) showed that PTSD patients had increased left amygdala activation during fear acquisition and decreased activity of the medial prefrontal cortex/anterior cingulate during extinction. It has been proposed that potentiating NMDA receptors using the glycine agonist
d-cycloserine may facilitate the extinction process when given in combination with behavioral therapy in patients with anxiety disorders
(189).