As much as 85% of the liability for bipolar disorder may be inherited
(1). Yet, although bipolar disorder has a strong genetic basis and some regions of the genome have been implicated, the genes involved in its etiology remain unknown
(2,
3). In fact, even when data from several genome scans were jointly examined by two different meta-analytic methods
(4,
5), discrepant findings of linked chromosomal regions emerged. Furthermore, in the more powerful of these two studies
(5), the best evidence for linkage at particular loci would be considered only suggestive according to traditional criteria of genome-wide statistical significance, despite a pool of over 1,000 affected individuals.
This uncertainty may be due, at least in part, to the clinical complexity and heterogeneity of the disorder. For example, although the DSM-III-R criteria and Research Diagnostic Criteria
(6) for bipolar disorder have excellent sensitivity and specificity in detecting a mental illness, the disorder may still be confused with major depression, schizophrenia, or schizoaffective disorder in as many as 10% of cases
(7). Furthermore, individuals with bipolar disorder differ markedly in illness severity and duration, rates of personal and familial suicidality and mood disorder, and extent of concomitant substance abuse and neuropsychiatric abnormalities. Such complexity creates classification problems that can restrict the power of genetic studies of the condition.
Some of the clinical heterogeneity of bipolar disorder may be due to pleiotropic effects of a single set of bipolar disorder genes; alternatively, such complexity could be due to underlying etiologic heterogeneity. This latter idea is supported by the fact that some multiply affected families show evidence for autosomal dominant transmission of a single major gene for bipolar disorder, while the segregation of the illness through other pedigrees is more consistent with a multifactorial and polygenic etiology in which numerous environmental and genetic factors interact
(8–
12). Clarifying the source of such discrepancies has become a high priority for those working to understand the true nature of bipolar disorder because of the promise that such an approach holds for refining the phenotype(s) of the illness and the subsequent identification of risk genes for particular disease subtypes, if they exist.
The age at onset of bipolar disorder has been considered as one potentially useful variable for constructing homogeneous groups of patients
(13,
14). Age at onset correlates with several clinical features, which could prove useful in explaining some of the clinical heterogeneity just discussed. An early age at onset is more often associated with a chronic course and a poorer response to mood stabilizers, while a later age at onset is associated with more severe abnormal thought content in patients selected for psychotic illness
(15). Age at onset may also be a useful marker of the degree of genetic contribution to disease development. For example, Grigoroiu-Serbanescu et al.
(16) found that different modes of transmission best fit the segregation of early- and late-onset bipolar disorder; early-onset forms were best explained by passage of a non-Mendelian major gene with a polygenic component, and late-onset forms showed multifactorial inheritance. Early-onset cases also show comorbidity and familial co-transmission with attention deficit and conduct disorders
(13,
17–20).
Because age at onset is both familial
(21) and heritable
(22), it is reasonable to use linkage analysis to detect genes that regulate this trait in bipolar families. However, the age at onset of bipolar disorder as defined by the age at which any affective symptoms first appear might be too broad a phenotype to be useful for genetic study. This phenotype comprises at least two potentially etiologically distinct features: age at onset of mania and age at onset of depression. Thus, to use a composite phenotype that includes both of these aspects of the illness may introduce some degree of causal heterogeneity, which may lower inferential power and hinder gene discovery. To overcome this potential limitation, we examined the heritability of distinct components of the age at onset of bipolar disorder, i.e., the age at onset of mania or depression in pedigrees through which bipolar disorder was segregating. Because the age at onset of depression was not significantly heritable, the remainder of this work focuses on the mania age-at-onset quantitative trait.
Discussion
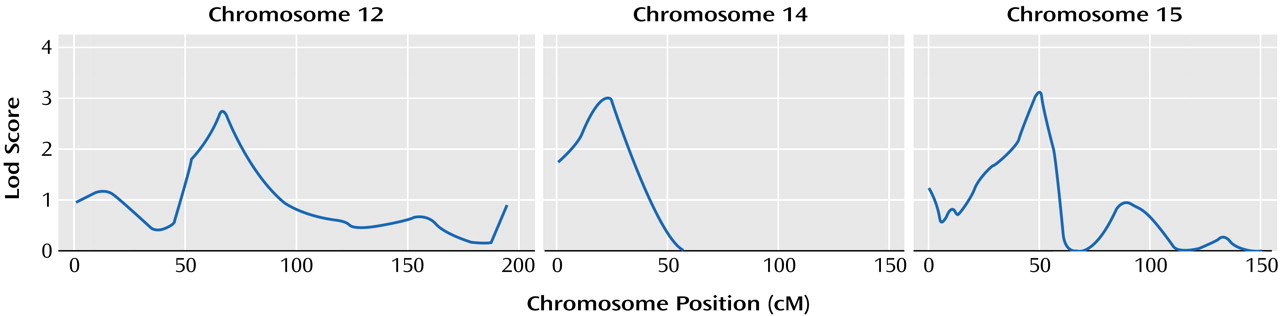
Our work suggests that three regions of the genome (chromosomes 12p, 14q, and 15q) may contain genes that influence the age at onset of mania in bipolar disorder. These chromosomal regions have not been implicated as high-risk loci for bipolar disorder in either of the previously published meta-analyses of bipolar disorder
(4,
5) or in previous linkage analyses of the current data set
(23–
26,
29,
30). This suggests that genes influencing the age at onset of mania in bipolar disorder are distinct from those influencing the liability of developing the disorder. Further studies are needed to confirm our finding and to determine whether these three loci act in concert—in either an additive or epistatic manner—to place individuals along a continuum of age at onset or whether these three loci are in some way associated with the three discrete age-at-onset classes delineated by Bellivier et al.
(39). Because these three regions of interest each span several megabases, it is premature to speculate about which genes gave rise to the observed linkage signals. However, these findings can serve as the basis for future fine-mapping efforts in these regions that may elucidate the responsible polymorphisms. It is clear that none of these linkage signals is attributable to the gene coding for the serotonin transporter (SLC6A4), which has been associated with age at onset of bipolar disorder
(40) but resides in chromosome region 17q11.1-q12. In addition, both of the genomic trinucleotide-repeat domains that have been associated with age at onset of bipolar disorder lie on chromosomes other than those showing the strongest linkage signals in the present study; ERDA1
(41) lies in chromosome region17q21.3 and CTG 18.1
(42) lies in region 18q21.1.
In addition to guiding subsequent work aimed at finding genes that determine age at onset generally, the present findings may also inform studies of distinct age-at-onset classes of the disorder, especially juvenile-onset bipolar disorder, which may be more genetically influenced than other forms of the illness. In fact, a growing body of evidence supports a greater genetic contribution to early-onset bipolar disorder than to later-onset forms of the disease
(13,
43).
For example, in the NIMH Collaborative Program on the Psychobiology of Depression, the age at onset of bipolar disorder in probands (range=13–52 years) was found to correlate inversely with the risk of the disorder for their relatives
(44). This relationship has also been demonstrated numerous times in studies that used a relatively late threshold (generally early or middle adulthood) for distinguishing between early- and later-onset bipolar disorder
(43); however, the effect of age at onset on familial risk is the most pronounced among the relatives of probands with pediatric onset (i.e., before 13 years)
(45). For example, Strober et al.
(46) found high rates of both bipolar disorder and major depression in the first-degree relatives of all of their patients with bipolar disorder but found a much higher prevalence of bipolarity in the relatives of pediatric patients with bipolar disorder (29.4%) than in relatives of older patients (7.4%). Similarly, Neuman et al.
(47) found that relatives of pediatric-onset bipolar patients were more than twice as likely to have bipolar disorder than were relatives of later-onset patients.
Such observations are consistent with a greater genetic influence on early-onset cases and suggest that pediatric cases may be more homogeneous, at least in their genetic etiology. If, as presumed, bipolar disorder has a multifactorial polygenic etiology in which numerous genes each have a small influence on total risk, then these genetically enriched early-onset cases of bipolar disorder may share a greater number of risk genes out of the total pool of responsible genes. For example, consider the hypothetical situation where bipolar disorder is caused by the combination of environmental risk factors with at least any four genes out of a pool of 10 risk genes. Among patients with a smaller genetic component (e.g., traditional cases of bipolar disorder), it may be common that the minimally sufficient number of risk genes (N=4) is possessed. Individuals in this category may share all four risk genes or, in fact, may have no overlap at all in the risk genes they possess. In an alternative scenario, among patients with a larger genetic component (e.g., patients with pediatric-onset bipolar disorder), it may be common for individuals to possess more than four of the necessary risk genes. The likelihood that one or more risk genes are shared by patients in this category (i.e., there is genetic overlap or homogeneity) increases with the number of risk genes that each individual possesses. Thus, if each individual in this category has six of the 10 total risk genes, some genetic overlap among patients is inevitable, as each case will have at least one risk gene in common with every other patient in the category; if each individual possesses all 10 risk genes, then the genetic overlap among them is absolute.
In addition to the greater risk for bipolar disorder it confers on relatives, pediatric-onset bipolar disorder is often characterized by an atypical clinical picture that often includes a chronic course, poor response to mood stabilizers, and high levels of comorbidity with attention deficit hyperactivity disorder and conduct disorder
(14,
48–52). It is interesting that this pattern is seen in only about one-third of adults with bipolar disorder
(53). In sum, these data suggest that individuals with an early onset of bipolar disorder are biologically different from those with a typical onset age and clinical presentation.
It is possible that the loci detected in our study of age at onset harbor genes that separate early-onset and later-onset cases. For example, these loci may contain genes whose risk alleles, if present, lead to an earlier onset of a more severe bipolar illness, while their absence merely allows the “typical” form of bipolar disorder to become manifest. This possibility cannot be powerfully examined in the present data set because we had no juvenile family members. As suggested by Todd et al.
(54), linkage studies of bipolar disorder would benefit from recruiting families ascertained through juvenile probands.
Several limitations to this study should be considered. First, although the group size in this study was moderate, the power to detect genes with small effects remains limited. Second, we did not account for other forms of genetic heterogeneity that could exist in this study group. Third, to limit the testing of multiple phenotypes and improve power, we considered only one definition of mania, which may not have been optimal.
In conclusion, the present study identified three chromosomal regions that may harbor genes regulating the age at onset of mania in families that are multiply afflicted with bipolar disorder. These genes appear distinct from those that may increase the overall susceptibility to the disorder. Our results provide suggestive evidence that the age at onset of mania is controlled by multiple quantitative trait loci, which further supports the idea that age at onset is a biologically meaningful feature of the disorder that may be useful in clarifying its heterogeneity.