Despite sharing a high 5-HT
2/D
2 binding ratio, the antipsychotic drugs we call “atypical” differ significantly from each other. In the case of risperidone and olanzapine, this “atypical” nature appears to be lost in a dose-dependent manner, resulting in the appearance of extrapyramidal side effects and sustained hyperprolactinemia at higher doses. Indeed, the relationship between dopamine D
2 receptor occupancy and clinical effects (response and extrapyramidal side effects) for risperidone and olanzapine in human subjects studied with PET is similar to that found with older antipsychotic drugs
(7). In contrast to risperidone and olanzapine, clozapine and quetiapine do not result in dopamine D
2 receptor occupancy approaching the threshold for extrapyramidal side effects, even when used at the higher end of their therapeutic dosing range
(2,
7,
8). This important difference cannot be reliably predicted from the respective affinities of these drugs for the 5-HT
2 and D
2 receptors. The ratio of 5-HT
2 receptor affinity to D
2 receptor affinity for risperidone, for example, is 10 times higher than that for quetiapine
(5,
6). Yet the in vivo separation of 5-HT
2 receptor occupancy compared with D
2 receptor occupancy is lower for risperidone than for quetiapine at clinical doses
(7,
8), demonstrating the critical importance of in vivo data obtained from patients being treated with antipsychotic drugs.
Ziprasidone is a novel antipsychotic drug that was approved for clinical use within the past few years. A benzothiazolylpiperazine, ziprasidone is structurally unlike other atypical antipsychotics, although clinically it too is characterized by a lower risk of extrapyramidal side effects and lack of sustained hyperprolactinemia
(9–
11). Similar to findings for the other atypical antipsychotics, in vitro data for ziprasidone indicate a pharmacological profile showing higher affinity for 5-HT
2 versus D
2 receptors
(12). Three previous PET studies have made separate evaluations of striatal D
2 receptor occupancy and frontal 5-HT
2 receptor occupancy by ziprasidone in healthy human subjects
(13–
15). These studies indicated that single doses of 20–40 mg of ziprasidone result in a dose-dependent dopamine D
2 receptor occupancy of >60% at 5 hours, while the cortical 5-HT
2 receptors are virtually saturated 4 hours after a single 40-mg oral dose. Although these studies provided preliminary data regarding the receptor binding profile of ziprasidone in vivo, their design did not allow for the study of the relationship between the 5-HT
2 and D
2 receptor binding profiles within the same subjects. Moreover, these data were obtained in studies of single-dose administration in healthy volunteers and did not fully explore the range of doses currently recommended for the treatment of psychosis.
Method
The study was approved by the Human Subjects Review Committee of the University of Toronto, and the subjects provided written informed consent after receiving detailed information about the protocol. Male and female patients were included if they were between the ages of 18 and 50 years, met DSM-IV criteria for either schizophrenia or schizoaffective disorder, and warranted a switch to ziprasidone, either because of side effects or lack of response in previous treatment. Subjects were excluded if they had had acute psychotic exacerbation within 3 months of the study or had a history of treatment resistance or of treatment with either clozapine within 3 months or a depot antipsychotic within 6 months. Subjects with a history of substance abuse within 3 months of the study, a positive urine drug screen, a history of a serious neurological or general medical condition, concurrent treatment with medications known to elevate the QTc interval, or current abnormalities on laboratory tests or ECG were also excluded. All subjects received a physical examination at baseline, as well as a 12-lead ECG and routine chemical and hematological laboratory studies.
On enrollment, subjects had a 2-day washout from previous antipsychotic treatment before random allocation to treatment with ziprasidone at one of four doses: 20, 40, 60, or 80 mg by mouth twice a day (for clarity, these groups will be referred to as the 40-, 80-, 120-, and 160-mg/day groups). The first two groups started treatment with 40 and 80 mg/day from day 1 and continued to receive this dose for the duration of the study. The 120- and 160-mg/day groups started treatment with 80 mg/day, and the dose was titrated to the target dose in an open-label manner between days 4 and 8. To minimize variance in absorption of the drug, subjects were instructed to take the medication with food
(10). The patients continued to take their target dose for at least 2 weeks to ensure steady-state plasma levels at the time of the PET studies. The following clinical rating scales were completed at baseline and at 3 weeks: Positive and Negative Syndrome Scale
(16), Simpson-Angus Rating Scale
(17), Clinical Global Impression
(18), and Barnes Rating Scale for Drug-Induced Akathisia
(19).
D
2 and 5-HT
2 receptor occupancy were assessed on the same day 12 and 14–16 hours, respectively, after the last administered dose. The [
11C]raclopride PET scans for D
2 receptor occupancy were obtained immediately after injection of 10 mCi of high-specific-activity [
11C]raclopride (>300 Ci/mmol) using a bolus plus infusion protocol
(20–
23), with 59% injected as a bolus over 1 minute and the rest injected by means of intravenous infusion over 74 minutes. After a brief transmission scan for attenuation correction of the emission scans, emission scans were obtained every minute for the first 15 minutes and then every 5 minutes until the end of the study at 75 minutes. The [
18F]setoperone PET scans for 5-HT
2 receptor occupancy were obtained after a bolus injection of 5 mCi of high-specific-activity [
18F]setoperone (>300 Ci/mmol)
(24,
25), with emission scans obtained every minute for the first 5 minutes and then every 5 minutes until the end of the study at 90 minutes.
PET scanning was conducted by using a brain-only GEMS PC2048-15B PET camera (General Electric Medical Systems, Milwaukee) that produced 15 6.5-mm-thick slices with a resolution of about 5–6 mm in air. Patients were scanned lying down and with fixation of the head achieved by using a thermoplastic face mask (Tru-Scan Imaging, Annapolis, Md.), allowing for repositioning between procedures.
To permit accurate delineation of the brain regions for data analysis, a magnetic resonance imaging (MRI) scan was done for each patient by using a GE Signal 1.5-T scanner (General Electric Medical Systems, Milwaukee). The image was acquired by using a conventional T
1 localizing scan and a fast spin echo sequence (both proton density/T
2 and T
1) with a 3-mm slice thickness. The MRI scan of each patient was coregistered to his or her PET scan by using RView8 software
(26). The regions of interest used in the analysis of D
2 and 5-HT
2 receptor occupancy were the caudate/putamen and frontal cortex, respectively, with the cerebellum used as a reference region for both receptor studies. The region-of-interest analysis was completed by using Alice 3.1 software (Perceptive Systems, Boulder, Colo.), which allows the rater to draw regions of interest on summed PET images (representing averaged images of the dynamic time series) coregistered to the subject’s MRI scan, which serves as an anatomical guide. The regions of interest were drawn by a single rater on two axial slices for the cerebellum (around the outermost border of cerebellar cortex), two axial slices for the striatum, and five axial slices for the frontal cortex (with the first level above the level of the orbit where the crown of the frontal cortex is established and the central sulci are clearly visible). The regions of interest were drawn such that their volume was larger than twice the full width at half maximum to minimize errors due to partial volume effects
(27). The regions of interest were then transferred to the dynamic PET images by using the same software, and a time activity curve was generated and used in the analysis.
D
2 and 5-HT
2 receptor binding potential was calculated by using previously described methods
(28–
30). For the D
2 receptor binding potential, the mean striatum/cerebellum ratio obtained between 30 and 75 minutes of scanning was used as an estimate of the equilibrium binding potential
(31). This timing was chosen on the basis of previous studies that showed a very high correlation between the binding potential derived from the ratio method and analytically derived estimates of D
2 binding potential (r>0.95)
(32). This method has been shown to be highly reliable, with a scan-rescan standard deviation of 6%, and it has been standardized in our lab with high intra- and interrater reliability (intraclass correlation coefficients: r>0.95)
(33). For 5-HT
2 receptor occupancy, we used the simplified reference tissue model to derive an estimate of the binding potential, with the cerebellum as the reference tissue and the frontal cortex as the region of interest
(34). Receptor occupancy for a given dose was then calculated as the percentage reduction of receptor binding potential with drug treatment, compared to baseline (100 × [1 – (binding potential
drug scan/binding potential
baseline)]). Age-corrected measures of binding potential were obtained from a previously collected data set of 13 (for D
2) and 20 (for 5-HT
2) antipsychotic-free healthy subjects. The data from these subjects were reanalyzed by the same rater who rated the study patients to ensure within-study consistency. The absence of the patients’ own baseline values introduced a potential error: for D
2 receptor occupancy this error, as calculated on the basis of variance in the data from antipsychotic-naive patients, was expected to vary from 0% to 9% for patients with 50% occupancy and from 0% to 4% for patients with 80% occupancy
(2,
32).
Venous blood was collected for measurement of ziprasidone and prolactin plasma levels at the time of the respective PET scans. The levels of ziprasidone were estimated in heparinized human plasma by using high-performance liquid chromatography with electrochemical detection (BAS Analytics, West Lafayette, Ind.). Prolactin levels were determined by using a two-site chemiluminometric immunoassay with a minimum detectable limit of 0.3 ng/ml and a coefficient of variance of 3.6%–4.5% (ACS, CIBA-Corning Diagnostics, East Walpole, Mass.).
Statistical analyses were carried out by using SPSS (SPSS, Inc., Chicago). Bivariate correlation analysis was used to examine the relationship between the primary variables of interest. Nonlinear regression analysis was used in the estimation of the plasma ziprasidone level associated with 50% receptor occupancy. Paired Student’s t tests were used to examine changes in laboratory and clinical variables over time between baseline and study completion.
Results
A total of 20 subjects were enrolled, and 16 completed the trial (mean age=33 years, SD=8; nine male and seven female subjects). Three patients received 40 mg/day of ziprasidone, three received 80 mg/day, five received 120 mg/day, and five received 160 mg/day. Of the four subjects who did not complete the protocol, two discontinued because of lack of medication efficacy, one discontinued because of severe somnolence/sedation, and one did not meet the inclusion criteria. Thirteen subjects had a diagnosis of schizophrenia, and the remaining three subjects had a diagnosis of schizoaffective disorder. All subjects had previously received the following antipsychotics as maintenance therapy: olanzapine (N=5), risperidone (N=5), quetiapine (N=2), and typical antipsychotics (N=4). Data from two [11C]raclopride PET scans (for one patient receiving 40 mg/day and one patient receiving 160 mg/day of ziprasidone) and two [18F]setoperone PET scans (for one patient receiving 40 mg/day and one patient receiving 80 mg/day of ziprasidone) could not be analyzed because of inadequate cerebellar coverage during image acquisition or technical difficulty with coregistration of the PET image with the MRI scans.
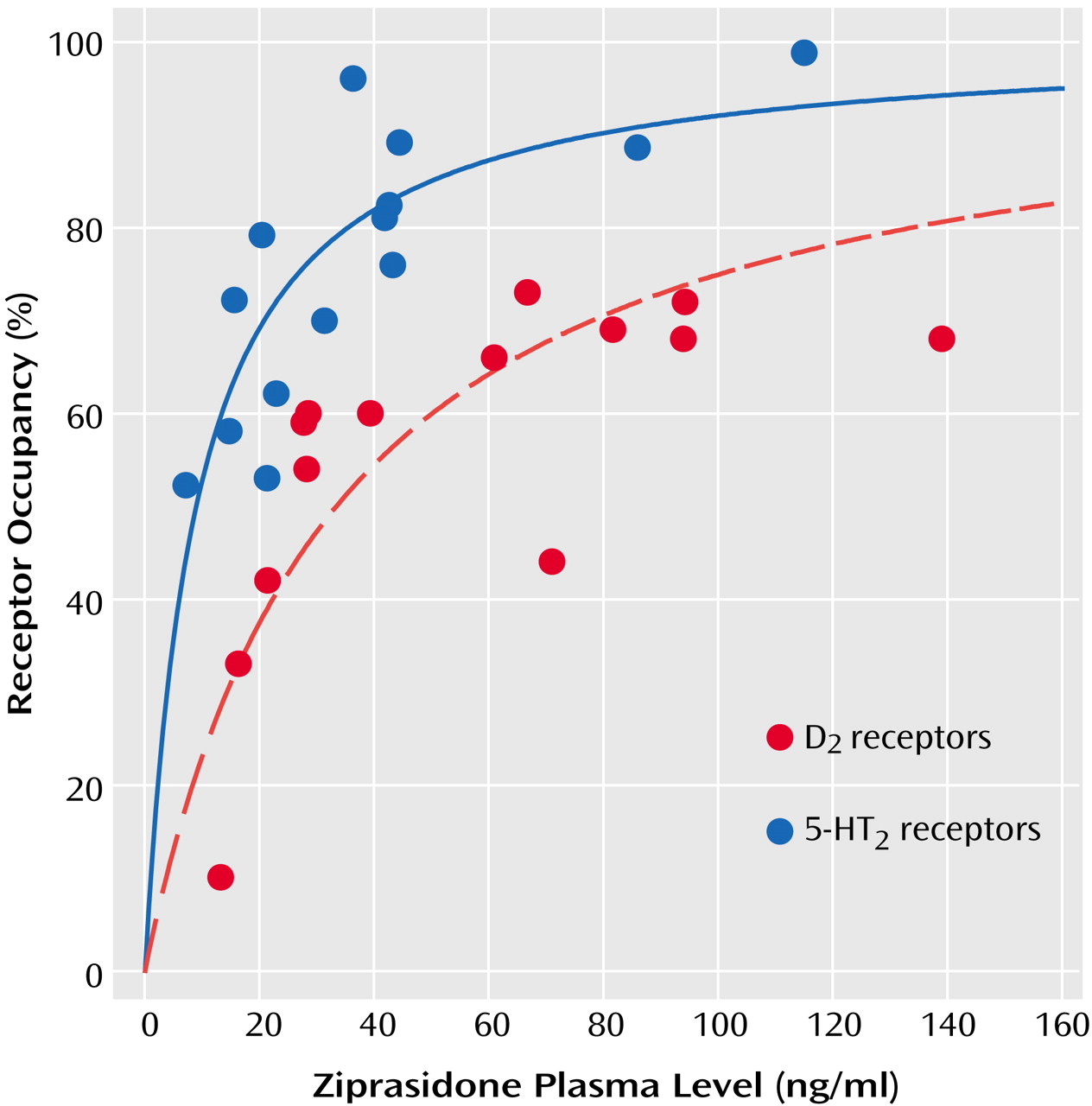
The mean D2 receptor occupancy was 56% (SD=18%, range=10%–73%), which was significantly lower than the mean 5-HT2 receptor occupancy (mean=76%, SD=15%, range=52%–99%) (t=3.14, df=26, p=0.04). The mean plasma ziprasidone levels were 53.4 ng/ml (SD=16.0) and 39.2 ng/ml (SD=30.2) at the time of the [11C]raclopride (12 hours after last dose) and [18F]setoperone (14–16 hours after last dose) PET scans, respectively. Ziprasidone dose was not significantly correlated with plasma levels at either time ([11C]raclopride PET scan: r=0.30, N=16, p=0.30; [18F]setoperone PET scan: r=0.30, N=16, p=0.20). Dose was related to 5-HT2 receptor occupancy (r=0.60, N=14, p=0.02) but not to D2 receptor occupancy (r=0.36, N=14, p=0.20).
Plasma level showed a significant positive correlation with both D
2 receptor occupancy (r=0.67, N=14, p<0.01) and 5-HT
2 receptor occupancy (r=0.75, N=14, p<0.002). As would be expected from the law of mass action determining bimolecular drug receptor interaction, the relationship between plasma ziprasidone levels and occupancy at both D
2 and 5-HT
2 receptors is better described by a saturation hyperbola (
Figure 1) conforming to the following equation: occupancy=a × [plasma level/(plasma level + ED
50)], where
a is the maximum receptor occupancy and
ED50 is the estimated plasma ziprasidone concentration (ng/ml) associated with 50% maximal receptor occupancy. The maximal mean occupancy (a) values calculated with this regression equation for D
2 and 5-HT
2 receptor occupancy were 84% (SE=10%) and 100% (SE=8%), respectively (
Table 1). Since the maximum D
2 receptor occupancy was not statistically different from 100%, and since the maximal expected D
2 receptor occupancy would be expected to be 100%, we constrained the equation for the relationship between D
2 receptor occupancy and plasma ziprasidone concentration so that a=100 (
Table 1 and
Figure 1). The estimated ED
50 for 5-HT
2 receptor occupancy (mean=9 ng/ml, SD=11) was almost fourfold lower than the ED
50 for D
2 receptor occupancy (mean=33 ng/ml, SD=49) (
Table 1).
There was a significant decline in the mean prolactin level at study completion, compared to baseline (mean change from baseline=–16.5 μg/liter, SD=16). This difference was primarily a result of normalization of previously elevated baseline levels in eight subjects. Only two subjects, both of whom were female subjects treated with 80 mg/day, developed elevated prolactin levels during ziprasidone treatment (an increase of 4 to 49 μg/liter and 27 to 31 μg/liter, respectively). The plasma ziprasidone level at the [11C]raclopride scan showed a modest correlation with prolactin level (r=0.65, N=16, p=0.006); however the prolactin level at this time was not significantly correlated with D2 receptor occupancy (r=0.39, N=14, p=0.17).
The medication itself was well tolerated in all but two individuals in the 120 mg/day group: one subject discontinued because of severe somnolence, and another subject developed oculogyric crisis. Since the study was designed to provide reliable PET data and not necessarily to detect clinical change, clinical data are provided here for complete description. Pooling the data across doses, there were nonsignificant decreases from baseline to endpoint in the mean total Positive and Negative Syndrome Scale score (mean=–4.9, SD=12.4) and the Clinical Global Impression severity scale (mean=–0.2, SD=1). Similarly, there was a small, nonsignificant decline over this same interval in extrapyramidal side effects and akathisia scores, as measured by the Simpson-Angus scale (mean=–0.6, SD=2) and Barnes scale (mean=–0.2, SD=2), respectively.
Given recent concerns about elongation of the QTc interval and increase in body weight with atypical antipsychotics, the results for these two variables obtained in this study will be reported in some detail.
The difference in the mean QTc interval before and after treatment was small (mean difference=3 msec, SD=28) (t=0.40, df=14, p=0.69). The number of individuals showing a decrease in the QTc interval was the same as the number showing an increase (N=8). In subjects with an increase, the ranges were as follows: 0–25 msec (N=4), 26–50 msec (N=2), and >50 msec (N=2). As a function of dose, the distribution of subjects showing an increase was as follows: none of three subjects who received 40 mg/day, two of three subjects who received 80 mg/day, three of five subjects who received 120 mg/day, and three of five subjects who received 160 mg/day. The two individuals with the increase of >50 msec (51 and 53 msec, respectively) were in the group that received 120 mg/day; only one individual had a QTc interval of greater than 450 msec, and this subject showed an increase of 51 msec (baseline: 407 msec; posttreatment: 458 msec).
There was no significant change in subjects’ mean weight at study completion (mean difference=–0.7 kg, SD=2.4). Four of the 16 subjects showed a mean weight gain of 2 kg (SD=1.4), while the rest of the subjects either showed no change in weight (N=4) or lost weight (N=8; mean=2.3 kg, SD=2.4, range=0.5–8.0) during the study.
Discussion
The principle result of this study is that ziprasidone shows a high ratio of 5-HT
2/D
2 receptor occupancy in vivo. This ratio is slightly underestimated since the [
18F]setoperone PET scans were completed 2–4 hours after the [
11C]raclopride scans. Our results are consistent with results from previous PET studies of healthy volunteers by Bench et al.
(13,
14) and Fischman et al.
(15). While Bench et al.
(14) did not provide estimates of ED
50, analysis of their published data showed that the estimated mean ED
50 for D
2 receptor occupancy (mean=15 ng/ml, SE=5) in their study does not differ significantly from that estimate in our study (mean=21 ng/ml, SE=7.5). Fischman et al.
(15) used a single-dose (40 mg), within-subject design to study 5-HT
2 receptor occupancy over time (4–18 hours) with [
18F]setoperone PET. Their results indicated saturation of the 5-HT
2 receptors at 4 and 8 hours after administration and a central half-life of 5-HT
2 receptor occupancy of almost 20 hours, which is consistent with the mean 5-HT
2 receptor occupancy calculated in this study (76% at 14 hours).
Visual inspection of the regression curve for dopamine D
2 receptor occupancy versus plasma levels (
Figure 1) suggests that the slope plateaus at around 65%–70% occupancy, which is above the threshold associated with optimal clinical response but below the D
2 receptor occupancy threshold that is associated with extrapyramidal side effects and prolactin elevation
(4). Although inspection of the curve may give the illusion of a “glass ceiling” for the D
2 occupancy of ziprasidone, we caution against such an interpretation. First, in a strict statistical sense, while the unconstrained maximal D
2 receptor occupancy was 84%, the 95% confidence interval included 100%, thus suggesting that maximal (i.e., 100%) occupancy cannot be ruled out. Second, since we used a relatively narrow dose range in this study (40–160 mg/day), we cannot rule out the possibility that occupancy may actually rise higher than 80% with higher doses. Finally, our estimates of occupancy were obtained 12 hours after the last dose. Ziprasidone reaches peak plasma concentrations within 6 hours of oral administration and has a relatively short half-life of 6–8 hours
(10). The single-dose data from Bench et al.
(14) indicate maximal occupancy of 75% within 4–8 hours after administration and a subsequent rapid decline to 50% 12 hours after the last dose.
The relatively high D
2 occupancies observed (maximum=73%) and the high maximum D
2 receptor occupancy predicted by the occupancy equation (mean=84%, SE=10%, which is not significantly different from 100%) suggest that ziprasidone’s occupancy profile bears greater similarity to that of risperidone and olanzapine than to that of clozapine or quetiapine. In a previous PET study comparing the binding profiles of risperidone, olanzapine, and clozapine (all measured 12 hours after administration of the last dose), the theoretical maximal D
2 receptor occupancy of risperidone and olanzapine was high (88% and 90%, respectively), compared to clozapine (68%)
(7). At therapeutic doses, the separation of ziprasidone’s 5-HT
2 and D
2 receptor occupancies in our study (
Figure 1) was 20%–30%, which is similar to that of risperidone and olanzapine (20%) but narrower than that of clozapine (>40%)
(7) and quetiapine (>40%)
(8). As we noted earlier, it is quite likely that if occupancy measures had been taken 6 hours after the last dose, 5-HT
2 and D
2 receptor occupancies would have been higher. However, since the 5-HT
2 occupancy was already close to ceiling, the effect on dopamine D
2 receptor occupancy would be more prominent.
Ziprasidone is indicated for the treatment of schizophrenia at a dose range of 40–160 mg/day. However the results from published clinical trials have been less consistent at lower doses
(9,
11), and these findings have been reflected in clinicians’ impression of limited clinical response in the lower dose range. Earlier impressions of efficacy at these lower doses came from PET data supported by a study by Bench et al.
(13), which suggested that an effective antipsychotic dose might be expected to be between 20 and 40 mg/day. Bench et al. found >60% D
2 receptor occupancy 5–6 hours (≈T
max) after the administration of a single oral dose of 20 mg (N=1) and 40 mg (N=1) of ziprasidone using an [
11C]raclopride bolus protocol in healthy volunteers. Our study addresses these methodological limitations through the use of multiple doses, scanning at steady state, the use of a bolus plus infusion schedule for [
11C]raclopride (which, compared with a bolus injection, approximates more closely the true equilibrium condition required for binding potential estimation), and the use of age-corrected measures of binding potential. We found that 60% D
2 receptor occupancy was not achieved until a mean plasma ziprasidone level of 50 ng/ml is reached (
Figure 1). Data from a study by Miceli et al.
(10) of the multiple-dose pharmacokinetics of ziprasidone under nonfasting conditions show that 120 mg/day is the minimum dose expected to result in serum ziprasidone levels equivalent to those in our current study (i.e., collected at 12 hours) that were associated with this therapeutic threshold. This minimum dose is consistent with the clinical impression that the optimal effective dose of ziprasidone is not 20–40 mg/day as originally suggested by Bench et al. but closer to 120 mg/day as suggested by our data.
We found no significant correlation between prolactin level at the time of [
11C]raclopride PET and D
2 receptor occupancy measured in the striatum. These results are at variance with several previous studies that have related striatal D
2 receptor occupancy to prolactin elevation
(4,
35–37), although the threshold of striatal D
2 receptor occupancy for prolactin elevation has varied between drugs. In a previous study of the relationship between occupancy, clinical response, and side effects in patients with first-episode schizophrenia treated with haloperidol
(4), we found that the likelihood of hyperprolactinemia associated with D
2 receptor occupancy <72% was only 15%. In the present study, none of the subjects had a D
2 occupancy >73%, which may in part account for the observed lack of correlation between prolactin level and D
2 receptor occupancy. Previously it was shown that these discrepant findings may be related to the fact that atypical antipsychotics have differential peripheral (pituitary) and central (striatal) D
2 receptor occupancy, which in turn is related to differential penetrability of a drug across the blood-brain barrier
(38,
39). Our finding of a low incidence of prolactin elevation with ziprasidone 12 hours after the last dose is consistent with the findings of Bench et al.
(14), who found that transiently elevated prolactin levels normalized in five of six subjects by 12 hours and in all subjects by 18 hours after administration of a single 40-mg dose of ziprasidone. Similarly, Miceli et al.
(10) found that this transient dose-independent increase in prolactin levels was maximal at 6 hours (T
max), consistent with recent studies showing a doubling of baseline prolactin levels within 6 hours of administration of antipsychotics in subjects treated with risperidone, olanzapine, quetiapine, and clozapine
(8,
40).
The current study is subject to limitations that are primarily related to the study design. Although the subjects were advised to take their medications with food at a particular time, the actual drug ingestion was not supervised and the study did not require a standardized diet. In contrast to a previous single-dose escalation study
(14), our study did not find a significant dose-related increase in D
2 receptor occupancy after 3 weeks of treatment. The main reason for this finding is that dose did not predict plasma levels, although we did find the expected curvilinear relationship between ziprasidone plasma level and receptor occupancy. These findings are likely related to the known effect of food on ziprasidone absorption
(41), which would result in relatively large between-subject variance in plasma levels for a given dose.
The design of the study permitted for occupancy evaluation at 12–16 hours, that is, at trough plasma ziprasidone levels, which is known to provide more stable and less variable estimates across subjects
(42,
43). Although this design allowed us to determine plasma occupancy relationships for D
2 and 5-HT
2 receptors, it did not permit the inclusion of higher plasma levels in the regression analyses, which increases the risk of underestimating maximal receptor occupancy and ED
50, particularly in the case of D
2 receptor occupancy. Furthermore, since ziprasidone shows very high plasma protein binding (>99%), minor fluctuations in protein binding would be expected to result in significant changes in the free ziprasidone levels available to cross the blood-brain barrier, further contributing to experimental error.
Finally, our study focused on striatal dopamine D
2 receptor occupancy. Several reports have drawn attention to the importance of extrastriatal dopamine D
2 receptor occupancy as an important variable
(44–
50). No systematic data are available regarding ziprasidone’s occupancy of extrastriatal dopamine D
2 receptors, and the relative contribution to antipsychotic efficacy of limbic striatal and extrastriatal (i.e., cortical) dopamine D
2 receptor occupancy remains unclear. Nonetheless, since it has been suggested that some atypical antipsychotics may show a differential dopamine D
2 receptor occupancy in the cortical regions, compared with the striatal regions
(44–
49), ziprasidone’s extrastriatal versus striatal D
2 receptor occupancy remains to be explored in future studies.
In summary, these results, to our knowledge, constitute the first study measuring the occupancy of therapeutically relevant doses of ziprasidone at D2 receptors and 5-HT2 receptors within the same subjects. The results provide in vivo evidence that ziprasidone shows a higher blockade of serotonin 5-HT2 receptors, compared to dopamine D2 receptors. Our PET data also suggest that the optimal effective dose of ziprasidone may be higher than that suggested by earlier studies. While ziprasidone’s 5-HT2/D2 receptor binding profile is consistent with that of other atypical antipsychotics (particularly risperidone and olanzapine), future studies that are focused on peak levels, that measure the plasma free fraction, and that examine extrastriatal occupancy in the context of a clinical study would help clarify the clinical importance of these occupancy data.