Generally, atypical antipsychotic drugs are better tolerated than typical antipsychotic drugs
(2). With the exception of clozapine, which has clear advantages for antipsychotic-resistant patients and suicidality
(2), these medications have offered, on average, only moderate advantages with respect to efficacy for positive and negative symptoms. However, they all show the ability to improve cognition, albeit only partially
(3). Nevertheless, the current group of atypical antipsychotic drugs produces less-than-optimal improvement in global measures of function such as quality of life and work and social function. For this reason, and because of a variety of metabolic and other side effects
(2), noncompliance remains a substantial problem. As a result, there is considerable interest in developing more effective and better tolerated classes of agents, preferably ones with truly novel mechanisms of action.
It has become increasingly clear that the pathophysiology, if not the etiology, of schizophrenia probably results from more than dopaminergic dysfunction
(4). Screening for novel compounds to treat schizophrenia has historically used the ability of compounds to block dopamine neurotransmission
(5). However, some models not directly targeting the dopamine system have also been used (e.g., blockade of phencyclidine-induced locomotor activity, prepulse inhibition, and the conditioned avoidance response). Using a combination of these strategies, we identified four novel compounds with unique mechanisms of action as potential antipsychotic agents. These include the following: 1) SR142801, a selective nonpeptide tachykinin NK
3 receptor antagonist; human NK
3 receptor K
i=0.22 nM in choline-containing cells
(6–
12). 2) SR46349B, a selective 5-HT
2A/2C receptor antagonist; human 5-HT
2A receptor IC
50=0.89 nM and 5-HT
2C receptor IC
50=10 nM in choline-containing cells
(13–
17). 3) SR141716, a selective antagonist for the central cannabinoid (CB
1) receptor; human CB
1 receptor K
i=5.6 nM in choline-containing cells and human CB
1 receptor K
i=17.5 nM in human substantia nigra tissue
(18–
21). 4) SR48692, a selective nonpeptide neurotensin (NTS
1) receptor antagonist; human NTS
1 receptor IC
50=8.7 nM in adult brain tissue
(22–
29).
A novel “meta-trial” design was developed for efficient, simultaneous initial evaluation of the therapeutic potential of these four compounds. Separate but identical protocols for each of these compounds were developed, each including haloperidol and placebo along with one investigational compound per protocol. An unbalanced random assignment method was used, and data from the groups receiving placebo and haloperidol from each of the studies were pooled and used to compare the efficacy and safety of each investigational drug. This allowed the use of a smaller total number of randomly assigned comparison patients; specifically, the protocol required fewer patients in the group receiving haloperidol and the group receiving placebo. We report here that two of the novel compounds studied, the NK3 and 5-HT2A/2C antagonists, had effects different from those of placebo but that the NTS1 and CB1 antagonists did not.
Method
Study Design
The meta-trial included four multicenter, double-blind, randomly assigned, parallel-group, placebo-controlled studies of four investigational compounds for the treatment of schizophrenia and schizoaffective disorder. Fifty participating centers in the United States were divided into six groups of centers (seven to 11 centers per group); two protocols were allocated to each group, and three groups of centers enrolled patients in each protocol. To ensure that data for all four investigational compounds were generated uniformly over time, each of the six groups of centers enrolled patients in four phases (20 patients per phase), alternating between the two protocols allocated to the groups. The protocols were approved by institutional review boards responsible for the participating centers, and written informed consent was obtained from each patient following a full explanation of study procedures.
Following screening and a 2- to 10-day single-blind placebo lead-in period, eligible patients were randomly assigned to receive once-daily treatment with either an investigational drug, haloperidol (10 mg/day), or placebo for 6 weeks in a 3:1:1 ratio. Doses of the investigational drugs were chosen on the basis of tolerability data in normal volunteers or effects on a pharmacodynamic measure (e.g., 18F-altanserin PET imaging of central 5-HT2 receptors for SR46349B) and were 200 mg/day for the NK3 antagonist, 5 mg/day for the 5-HT2A/2C antagonist, 20 mg/day for the CB1 antagonist, and 180 mg/day for the NTS1 antagonist.
All psychotropic medications and medications for the treatment of extrapyramidal symptoms were discontinued during the lead-in period. Agitation was treated with lorazepam at doses no greater than 6 mg/day during the lead-in period and the first week of randomly assigned treatment, and no greater than 4 mg/day during the remaining 5 weeks of randomly assigned treatment. Insomnia was treated with chloral hydrate (500–2000 mg/day) or lorazepam (maximum 2 mg/day). Benztropine (maximum 2 mg b.i.d.) was used to treat extrapyramidal symptoms if needed.
Patient Selection
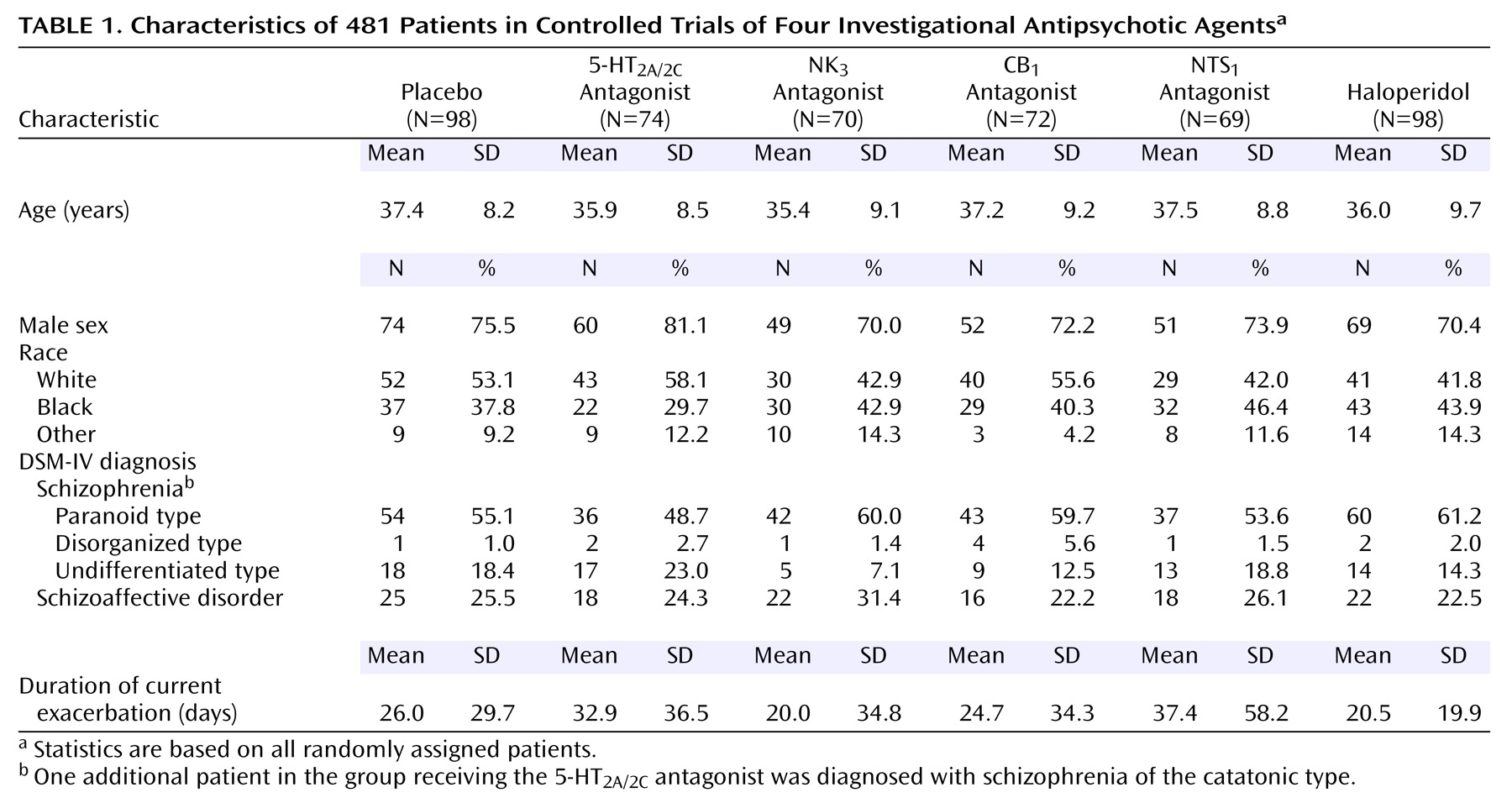
Men and women 18 to 64 years old who had schizophrenia or schizoaffective disorder diagnosed according to DSM-IV criteria were eligible for the study. Patients were required to be hospitalized at baseline through day 15 after random assignment to treatment. Eligible patients were also required to have a total score on the Positive and Negative Syndrome Scale
(31) greater than 65 at screening and baseline, including a minimum score of 4 (moderate) on at least two of four Positive and Negative Syndrome Scale positive symptom items (delusions, conceptual disorganization, hallucinatory behavior, and suspiciousness/persecution). A minimum severity of illness score of 4 (moderately ill) on the Clinical Global Impression (CGI)
(32) at screening and baseline was also required. Patients who recently received a depot antipsychotic were required to be free of that antipsychotic for at least one cycle preceding baseline.
Patients with other axis I DSM-IV diagnoses were excluded from the study, as were patients considered by the investigator to have been nonresponsive to treatment with at least two different classes of antipsychotic medications, patients with any clinically significant medical illnesses, patients with clinical laboratory or ECG abnormalities, patients with evidence of current substance abuse or dependence, and patients who were a danger to themselves or others.
Assessments
Assessments based on the Positive and Negative Syndrome Scale, CGI, and Calgary Depression Scale
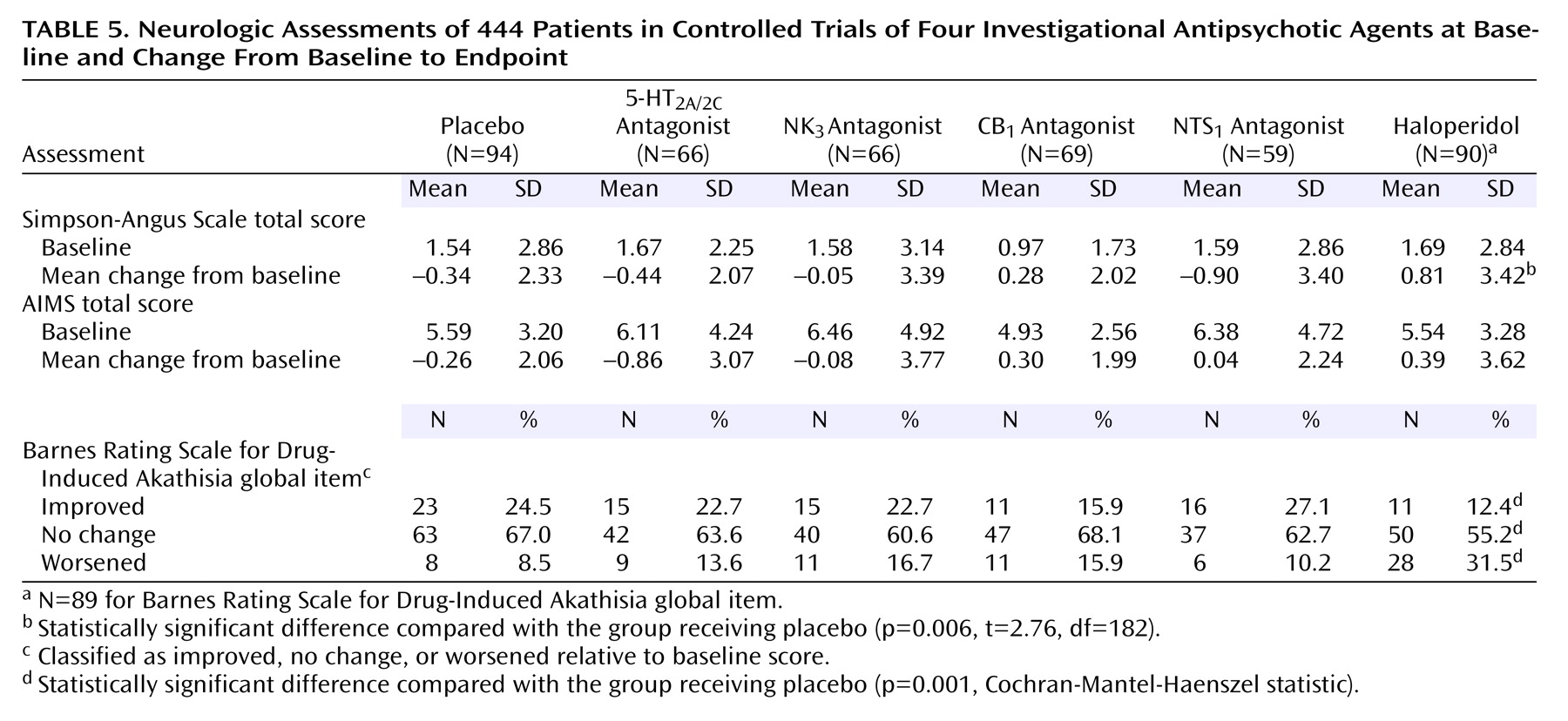
(33) were conducted at screening, baseline, 4 days after random assignment to treatment, and weekly thereafter during the 6-week double-blind treatment period. Safety assessments included spontaneously reported adverse events and measurements of vital signs and weight at each scheduled efficacy evaluation; evaluation of extrapyramidal symptoms based on the Simpson-Angus Rating Scale
(34) and the Barnes Rating Scale for Drug-Induced Akathisia
(35) at baseline and weeks 1, 2, 3, 4, and 6; evaluation of involuntary movements based on the Abnormal Involuntary Movement Scale (AIMS)
(36) at baseline and week 6; clinical laboratory tests at screening, baseline, and weeks 1, 3, and 6; 12-lead ECG at screening, baseline, and weeks 3 and 6; and physical examination at screening, baseline, and week 6. Blood samples to determine plasma drug levels were obtained on a weekly basis during the double-blind treatment period, approximately 12 hours after the most recent drug or placebo dose.
Data Analysis
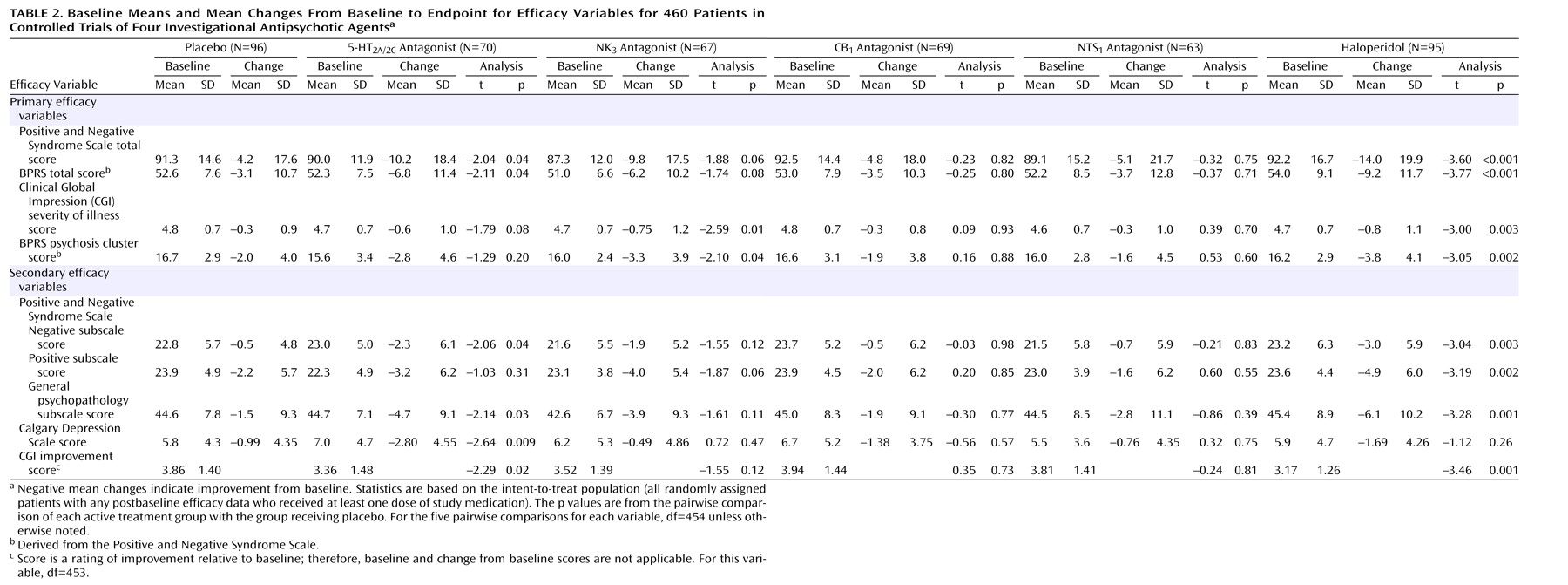
The analysis of efficacy was based on the intent-to-treat population, defined as all randomly assigned patients who received at least one dose of study medication or placebo and provided at least one postbaseline efficacy evaluation while receiving the medication or placebo. The primary time point was week 6. For patients who withdrew from the study before week 6, the last observation was carried forward and used in the primary analyses of efficacy. The primary efficacy variables were the changes from baseline to week 6 in the Positive and Negative Syndrome Scale total score, CGI severity of illness score, Brief Psychiatric Rating Scale (BPRS)
(37) total score (derived from the Positive and Negative Syndrome Scale), and BPRS psychosis cluster score (derived from the four Positive and Negative Syndrome Scale positive symptom item scores). Secondary efficacy variables included the changes from baseline to week 6 in the Positive and Negative Syndrome Scale negative, positive, and general psychopathology scores and Calgary Depression Scale total score. The CGI improvement score at week 6 was also a secondary efficacy variable.
One-way analysis of variance (ANOVA), with treatment group as a factor, was used to analyze all efficacy variables. By virtue of the statistical properties of the design of the trial (i.e., incomplete block design) we are assured that the estimated treatment effect is independent of any center-to-center variation. This is further corroborated by the following empirical evidence in the study: 1) the primary efficacy endpoint scores of the placebo and haloperidol treatments were similar across groups of centers, and 2) the differences between the group receiving placebo and the group receiving haloperidol among groups of centers and studies were consistent. In an attempt to quantify our claim, we also tested for the center group and treatment-by-center-group interaction using an ANOVA model. The p values were not statistically significant (all p values >0.10) and, therefore, were removed from the final ANOVA models. Planned pairwise comparisons were based on least-squares means from this model (with type III sums of squares) and included comparisons of each investigational drug group with the group receiving placebo as well as comparisons of the group receiving haloperidol with the group receiving placebo. The group receiving haloperidol was included strictly as an internal standard; therefore, no formal comparisons between this group and the investigational drug groups were made. Pairwise comparisons used two-sided tests at the 5% level with no adjustment for multiple comparisons.
Analysis of covariance (ANCOVA), including the baseline score and treatment group as factors, was used to conduct exploratory analyses of the primary efficacy variables.
A nonlinear regression model was used to investigate the relationship between the change from baseline in BPRS total score and median plasma concentration at endpoint for the two compounds demonstrating efficacy—the NK3 and 5-HT2A/2C antagonists.
The analysis of safety data was based on all randomly assigned patients who received at least one dose of study medication or placebo. Treatment-emergent adverse events were assigned preferred terms according to World Health Organization adverse reaction terminology. Incidence rates were calculated for each preferred term by treatment group, and Fisher’s exact tests were used to conduct pairwise comparisons of active treatment groups with the group receiving placebo. A one-way ANOVA model including treatment group was used to analyze changes from baseline to endpoint in the Simpson-Angus Scale and AIMS total scores. The Cochran-Mantel-Haenszel row mean scores statistic was used to analyze the proportions of patients whose scores on the Barnes Rating Scale for Drug-Induced Akathisia global item improved, showed no change, or worsened from baseline endpoint. A one-way ANOVA model with two-sided tests at the 10% significance level was used to analyze changes from baseline to endpoint in vital signs, clinical laboratory tests, and ECGs.
Sample size requirements for this study were based on a minimum of 80% power to detect a difference of 5 points between a group receiving an investigational drug and the group receiving placebo in the mean change from baseline in Positive and Negative Syndrome Scale total score, with the assumption of a within-group standard deviation of 10 points and a two-sided test at the 5% significance level. A sample size of at least 420 patients, therefore, was required, including 63 patients in each of the four investigational drug groups, 84 in the group receiving haloperidol, and 84 in the group receiving placebo.
Discussion
The major results of this trial are that the novel concept of a meta-trial to compare efficacy and tolerability of multiple, novel antipsychotic compounds simultaneously was validated and that two of the compounds, an NK3 antagonist (SR142801) and a 5-HT2A/2C antagonist (SR46349B), have sufficient activity, albeit in somewhat different domains, to warrant further study.
The novel meta-trial design was developed as an alternative, more efficient initial evaluation of the therapeutic potential of these four compounds. The 3:1:1 random assignment in each individual protocol and the incomplete block design of the study, with consistent placebo and haloperidol effects at each center, allowed an adequate number of patients to receive each of the investigational compounds and minimized the number of patients receiving haloperidol and placebo without compromising power.
The group studied comprised patients with moderate to severe symptoms of schizophrenia or schizoaffective disorder responsive to previous antipsychotic therapy. The consistent treatment effects observed in patients treated with haloperidol demonstrated that patients enrolled in the study were, on average, responsive to conventional drug therapy
(38–
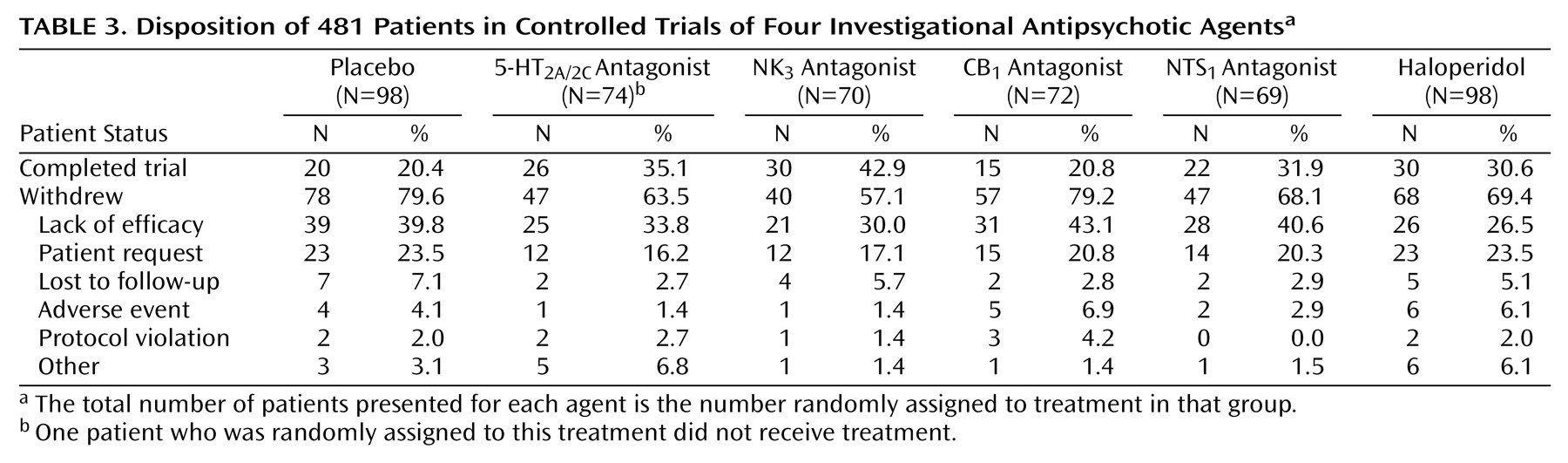
40). The modest size of these effects suggests that some patients may actually have been partially or poorly responsive and may have been chronically symptomatic rather than acutely exacerbated. Additionally, the high dropout rate noted across treatment groups, although consistent with other placebo-controlled trials in such patients, likely contributed to an underestimation of the true treatment effects.
Clinical improvement determined by scores on several rating scales was demonstrated for the 5-HT
2A/2C and NK
3 receptor antagonists. In accord with its ability to increase prefrontal cortical dopamine release
(17), significant differences in the group receiving the 5-HT
2A/2C antagonist were seen in measures of global, nonpsychotic symptoms, negative symptoms, and depression. Significant differences in the group receiving the NK
3 antagonist were seen in global measures and in measures of positive symptoms. In both groups, the treatment effects were smaller than those seen in the haloperidol-treated group.
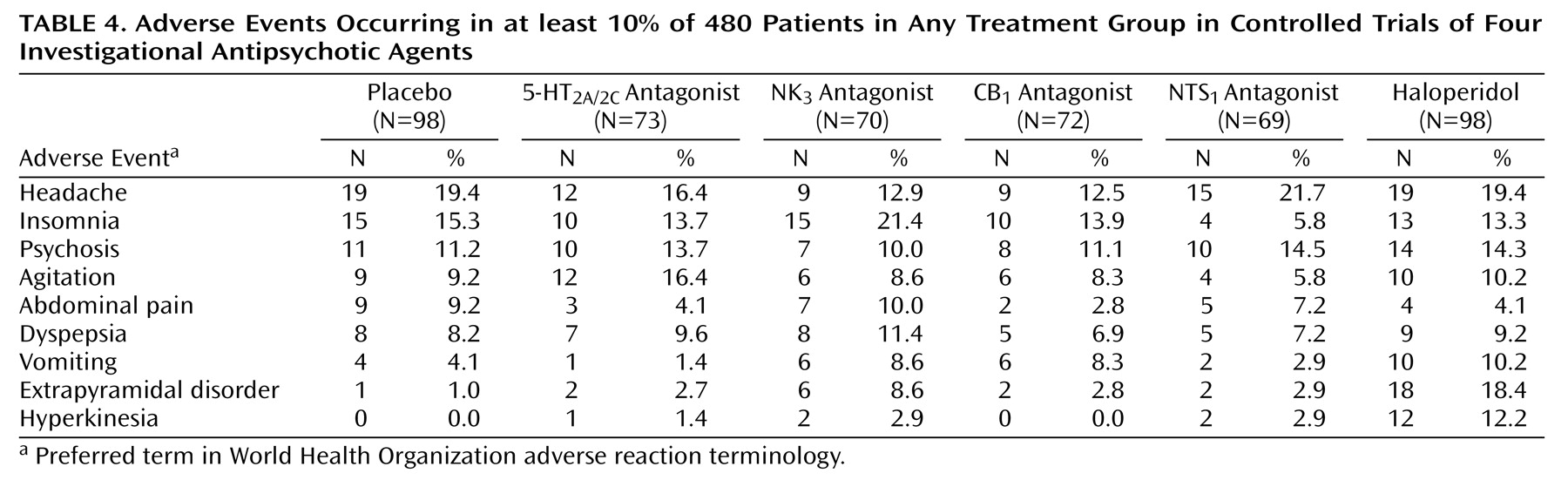
Both the 5-HT2A/2C and the NK3 receptor antagonists were well tolerated; no major safety issues arose. Of particular interest was the low risk of extrapyramidal symptoms and weight gain with both of these drugs.
Both clozapine and olanzapine have potent 5-HT
2A/2C antagonist properties, as well as many other potent actions on other receptors and transporters
(1). Risperidone is a weak 5-HT
2C antagonist
(41). None of these three compounds is a potent NK
3, NTS
1, or CB
1 antagonist. In vivo studies in rodents suggest a possible basis for the ability of a selective 5-HT
2A/2C antagonist such as SR46349B to improve total pathology and, in particular, negative symptoms in patients with schizophrenia. Clozapine, olanzapine, and risperidone, all of which are 5-HT
2A and D
2 receptor antagonists, preferentially enhance dopamine release in the rat medial prefrontal cortex
(42–
44). This effect is related to combined 5-HT
2A and D
2 receptor blockade
(44). Increased release of dopamine in the cortex may be expected to improve cognition, negative symptoms, and, perhaps, depressive symptoms
(44,
45). SR46349B, at 10 mg/kg but not 1–3 mg/kg, by itself can increase dopamine release in the medial prefrontal cortex without increasing dopamine release in the nucleus accumbens
(17). SR46349B (3 mg/kg) also potentiated haloperidol-induced dopamine release in both regions
(17). WAY100635 (0.2 mg/kg), a 5-HT
1A antagonist, abolished the effects of haloperidol plus SR46349B on dopamine release in the medial prefrontal cortex but did not in the nucleus accumbens
(17). The effects of WAY100635 on SR46349B- and clozapine-induced dopamine release in the cortex are not significantly different
(46). These results suggest that SR46349B-induced 5-HT
2A/2C antagonism may be advantageous alone or as an adjunct to D
2 antagonists to improve cognition and negative symptoms in schizophrenia. M100907 has been reported to have some efficacy as monotherapy in the treatment of schizophrenia
(47). It remains to be determined if both SR46349B and M100907 would be effective as monotherapy in some patients with psychosis or whether they would potentiate the effect of D
2 antagonists such as haloperidol or amisulpride, which lack 5-HT
2A/2C antagonist properties at clinical doses
(17).
Demonstration of efficacy with the NK
3 antagonist was the most interesting finding. Several lines of evidence suggest that NK
3 antagonists may be effective in the treatment of schizophrenia. NK
3 receptors are located in brain regions implicated in the pathophysiology of schizophrenia, including frontal, temporal, and parietal cortices as well as the striatum, substantia nigra, and hippocampus. NK
3 antagonists have been shown to modulate the activity of dopamine neurons in the ventral tegmentum and the pars compacta of the substantia nigra
(11,
12). Finally, NK
3 antagonists blocked the conditioned avoidance response in guinea pig (data on file at Sanofi-Synthelabo Research).
The lack of effect of the selective antagonist for the central CB
1 receptor SR141716 and the selective nonpeptide NTS
1 receptor antagonist SR48692 may be due to an inadequate dose, failure of these drugs to penetrate the blood-brain barrier in sufficient concentration, or lack of clinical activity of compounds with these mechanisms of action. The results with the CB
1 receptor antagonist are disappointing because a growing literature suggests a role for the CB
1 receptor in schizophrenia
(48–
50). Finally, Binder et al.
(51,
52) suggested that neurotensin agonists, rather than antagonists, may be effective antipsychotic drugs and that diminished neurotensin activity might be involved in the pathophysiology of schizophrenia.
In conclusion, results from this preliminary study allowed the identification of two investigational compounds, an NK3 antagonist and a 5-HT2A/2C antagonist, neither of which have significant D2-receptor-blocking properties, as candidates for further development as antipsychotic agents, alone or as adjunctive treatment, for schizophrenia, schizoaffective disorder, and perhaps other psychoses such as those associated with bipolar disorder, psychotic depression, or organic disorders such as Alzheimer’s disease and Parkinson’s disease. Additional studies are required to confirm the efficacy of these novel compounds and to explore dose-response relationships and characterize tolerability profiles more fully.