Despite the introduction of several new antipsychotics such as olanzapine, risperidone, and quetiapine—which share clozapine’s advantageous profile of a relatively low risk for extrapyramidal symptoms and no prolactin elevation—clozapine remains unique in its efficacy in the treatment of refractory schizophrenia
(1–
5). It has been established that patients with treatment-resistant schizophrenia may still improve from clozapine after nonresponse to the novel antipsychotic olanzapine
(6), although one study reported equal efficacy of olanzapine and clozapine in otherwise treatment-resistant schizophrenia
(7). Clozapine has also been noted to be superior to risperidone in patients suffering from severe chronic schizophrenia with poor previous treatment response
(8) and in chronically hospitalized patients with treatment-resistant schizophrenia
(9). In a study that used less stringent criteria for treatment resistance and that had a surprisingly high response rate to both risperidone and clozapine, risperidone reportedly showed equal efficacy to clozapine
(10). Up to now no study to our knowledge has been carried out to compare clozapine with quetiapine in patients with treatment-resistant schizophrenia—although the general impression is that quetiapine is not especially effective in treatment-resistant cases. Thus, clozapine still remains the most effective antipsychotic in cases of schizophrenia otherwise unresponsive to treatment.
Various explanations have been put forward in an attempt to characterize clozapine’s pharmacological uniqueness, which include its affinity for the dopamine D
4 receptor
(13), its potent serotonin 5-HT
2A receptor antagonism
(14), and its robust alterations of noradrenergic biochemistry
(15,
16). In addition, a hypothesis has been formulated stating that moderate occupancy of D
2 receptors might be clinically superior to a more complete D
2 blockade
(17). None of these theories have held so far. In a double-blind, controlled study of 97 patients, the potent D
4 and 5-HT
2A receptor antagonist fananserin proved to be an ineffective antipsychotic
(18). Further, the moderate D
2 occupancy hypothesis was rejected because patients who had been receiving oral neuroleptics and who were switched to clozapine did not differ from patients who had been receiving depot medications
(19). In the oral discontinuation group, one could expect to find a faster decline in D
2 occupancy, and if moderate D
2 blockade were the key to clozapine’s uniqueness, this group should have shown a more rapid response, which was not the case.
Clozapine’s relatively high affinity for the dopamine D
1 receptor may be related to its unique clinical efficacy, as D
1 receptors mediate the reward function in animal models, a principle thought relevant for the therapeutic action of antipsychotics
(20). In vitro, clozapine shows a relatively high affinity for D
1 receptors together with a moderate affinity for D
2 receptors
(21,
22). In man, clozapine showed a distinctively lower D
2 occupancy and a higher D
1 receptor occupancy compared with the typical neuroleptics
(23), but no comparisons with the newer atypical antipsychotics are available.
Method
We included 25 patients (18 men and seven women; mean age=35.4 years, range=18–58) with a DSM-IV diagnosis of schizophrenia or schizoaffective disorder in the study. Their diagnosis of schizophrenia was ascertained with the Structured Clinical Interview for DSM-IV, which was administered by an experienced psychiatrist (J.T., N.P.L.G.V., or O.A.). We recruited the patients from the Schizophrenia and Continuing Care Program of the Centre for Addiction and Mental Health in Toronto, where each patient received ongoing antipsychotic medication either as an inpatient or outpatient. All subjects gave their written consent after the procedure had been fully explained. The study and recruitment procedures were approved by the Research Ethics Board of the Centre for Addiction and Mental Health and the University of Toronto.
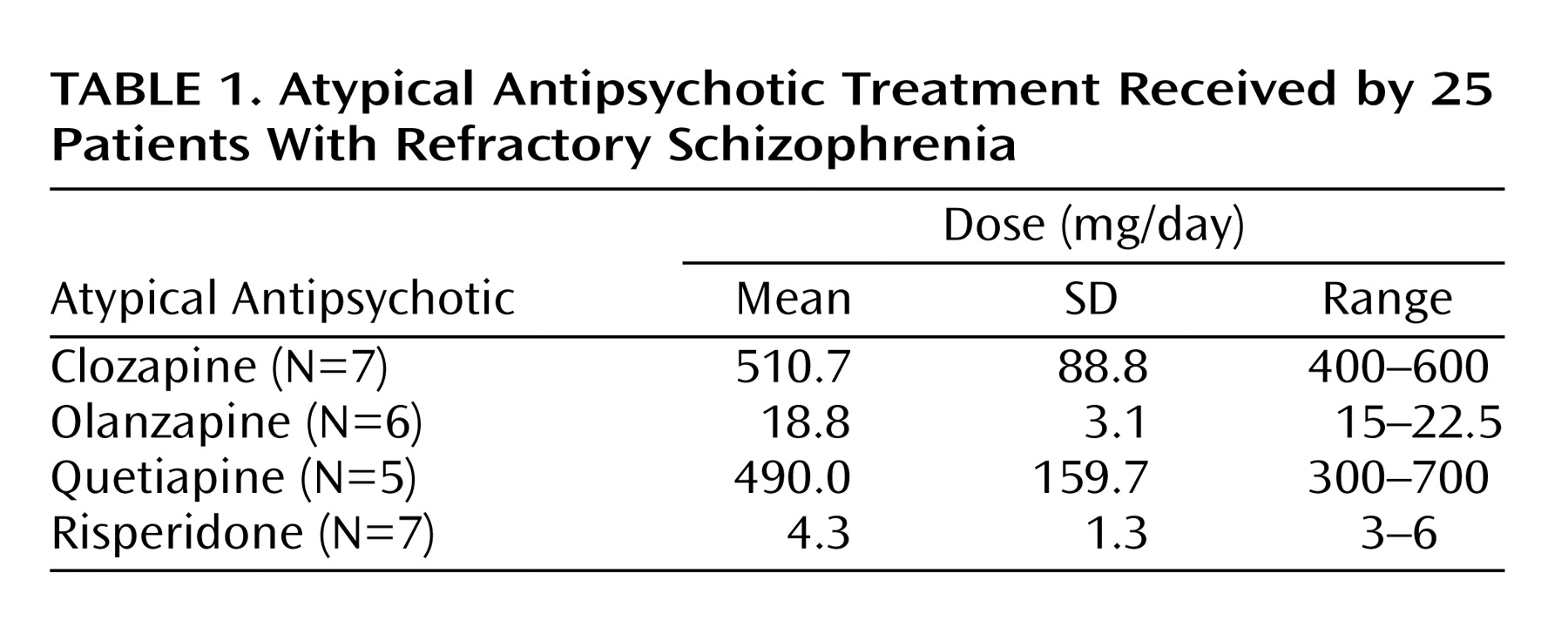
Patients had been receiving atypical antipsychotic treatment for at least 14 days before the PET study. A summary of the treatment regimens received by the patients is presented in
Table 1. In general, medications were prescribed once daily, twice daily if that was not tolerated. At the day of the PET scans, all patients took their total daily dose of medication at once approximately 2 hours before the first PET scan, and their striatal D
1 and D
2 occupancy were measured subsequently.
PET Scanning Procedures
Striatal dopamine D
2 receptor occupancy was determined by using 10.8 mCi (SD=2.7) of high-specific-activity [
11C]raclopride (mean=692.9 Ci/mmol, SD=465.0) administered as a bolus plus continuous infusion. Striatal D
1 receptor occupancies were determined by using 9.9 mCi (SD=0.6) of high-specific-activity [
11C]SCH23390 (mean=875.7 Ci/mmol, SD=328.8) administered as a bolus. Imaging was performed with a Scanditronix/GEMS PC-2048–15B head scanner as described in previous publications
(24–
27). The PET scans were performed in a fixed order starting with the [
11C]SCH23390 scan approximately 2 hours after the last dose and continuing with the [
11C]raclopride scan approximately 5–6 hours postdose.
A magnetic resonance imaging (MRI) scan was obtained for each of the patients (GE Signa 1.5-T scanner, proton density maps) and was coregistered to the composite [
11C]raclopride and [
11C]SCH23390 PET scans by using RView8/mpr software
(28). As described in previous publications
(24–
27), we drew the striatal (caudate plus putamen) and cerebellar regions of interest on two contiguous PET slices with reference to the overlapping coregistered MRI scan. The cerebellar time-activity curve was taken as an estimate of the free and nonspecific [
11C]raclopride binding
(29), while the striatal time-activity curve provided an estimate of specific binding to the D
2 receptors plus free and nonspecific binding. Under these assumptions, it can be shown that the striatal-cerebellar ratio minus one, at the time when the binding is at equilibrium (30–75 minutes in the aforementioned scans), provides an index proportional to the B
max/K
d ratio of [
11C]raclopride for dopamine D
2 receptors (referred to as the binding potential). In previous studies
(30) we have demonstrated that this ratio method correlates very well (r>0.95) with analytically derived estimates of D
2 binding potential, is highly reliable with a scan-rescan standard deviation of 6%, and has been standardized in our laboratory with excellent inter- and intrarater reliability (intraclass correlation coefficients >0.95). For estimation of specific binding to D
1 receptors (plus free and nonspecific binding) in the striatum and frontal cortex, a ratio was calculated between those regions and the cerebellum as a reference region
(27). Data from the left and right hemispheres were pooled for all subsequent calculations, since there was no significant asymmetry in D
1 or D
2 binding potentials.
Since we did not have baseline measures of D1 or D2 binding potentials for the patients, we used an age-corrected estimate from a comparison group of 29 untreated healthy volunteers who had no current or past axis I DSM-IV psychiatric disorder as determined with the Structured Clinical Interview for DSM-IV, nonpatient version, and had not taken any psychotropic medication in the 3 months preceding this study. Twelve subjects (five men and seven women) with a mean age of 32 years (SD=11, range=20–49) served as the comparison group for D1 binding potential, and 17 subjects (nine men and eight women) with a mean age of 29 years (SD=6, range=20–40) served as baseline D2 binding potential values.
Determination of Drug and Prolactin Plasma Levels
At the time of the PET scans, blood was drawn for a plasma drug level and prolactin level analysis. We determined clozapine, olanzapine, quetiapine, and risperidone levels in heparinized plasma using a liquid chromatography/mass spectroscopy method
(31,
32). Prolactin levels were determined by using a two-site chemiluminometric immunoassay with a minimum detectable limit of 0.3 ng/ml and a coefficient of variance of 3.6% to 4.5% (ACS, Ciba-Corning Diagnostics, Corning, N.Y.).
Statistical Analysis
Statistical analyses were performed with SPSS for Windows 11.0.1 (SPSS, Inc., Chicago, 2001). Dopamine D1 and D2 receptor binding indices of different antipsychotics were compared by using a general linear model (univariate analysis of variance [ANOVA]) with post hoc Scheffé tests for differences between clozapine and other atypical antipsychotics. Putative relations between plasma prolactin levels and D1 or D2 occupancies, respectively, were calculated by using Pearson’s product-moment correlation coefficients. All tests were performed two-tailed with an alpha level of p<0.05 set as the threshold for statistical significance.
Results
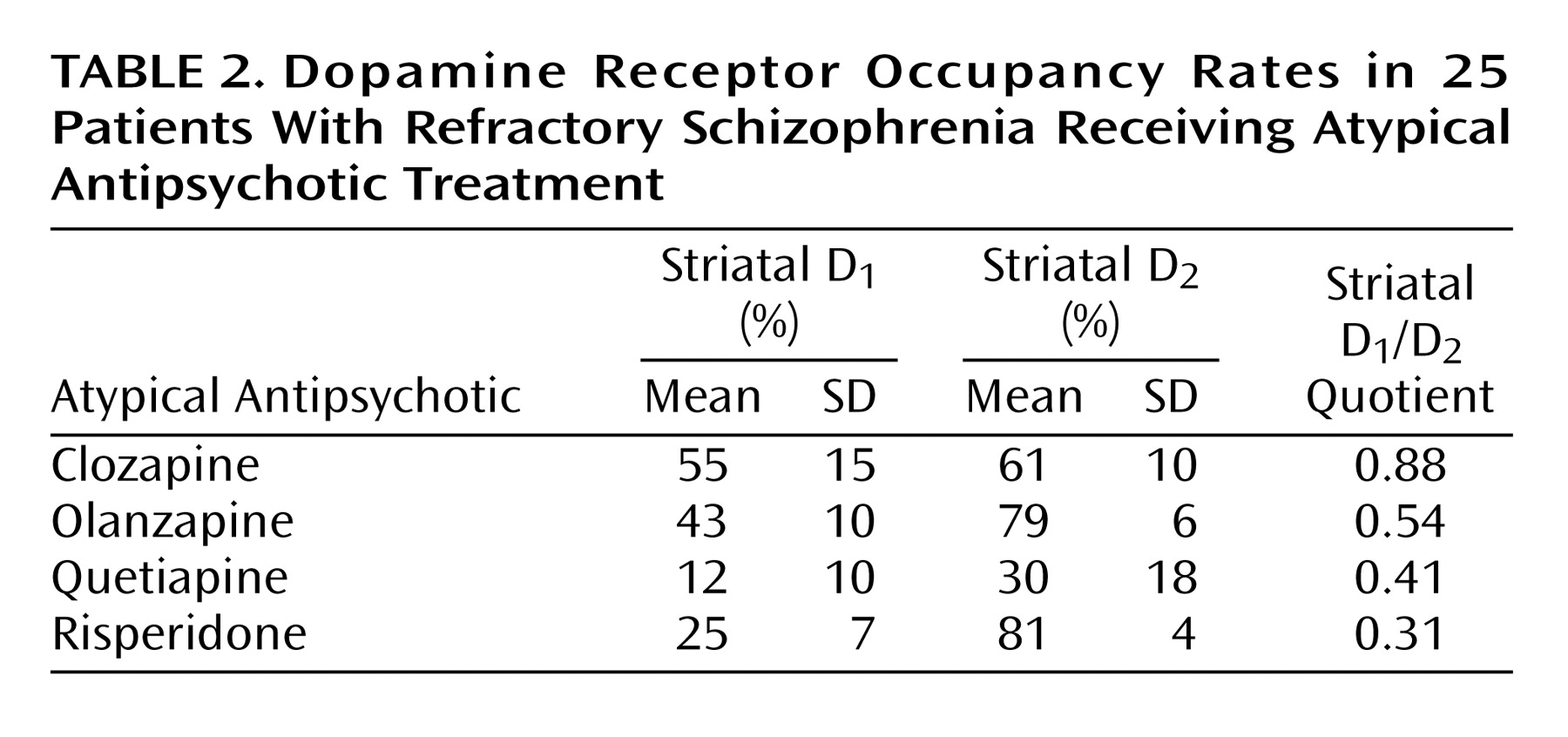
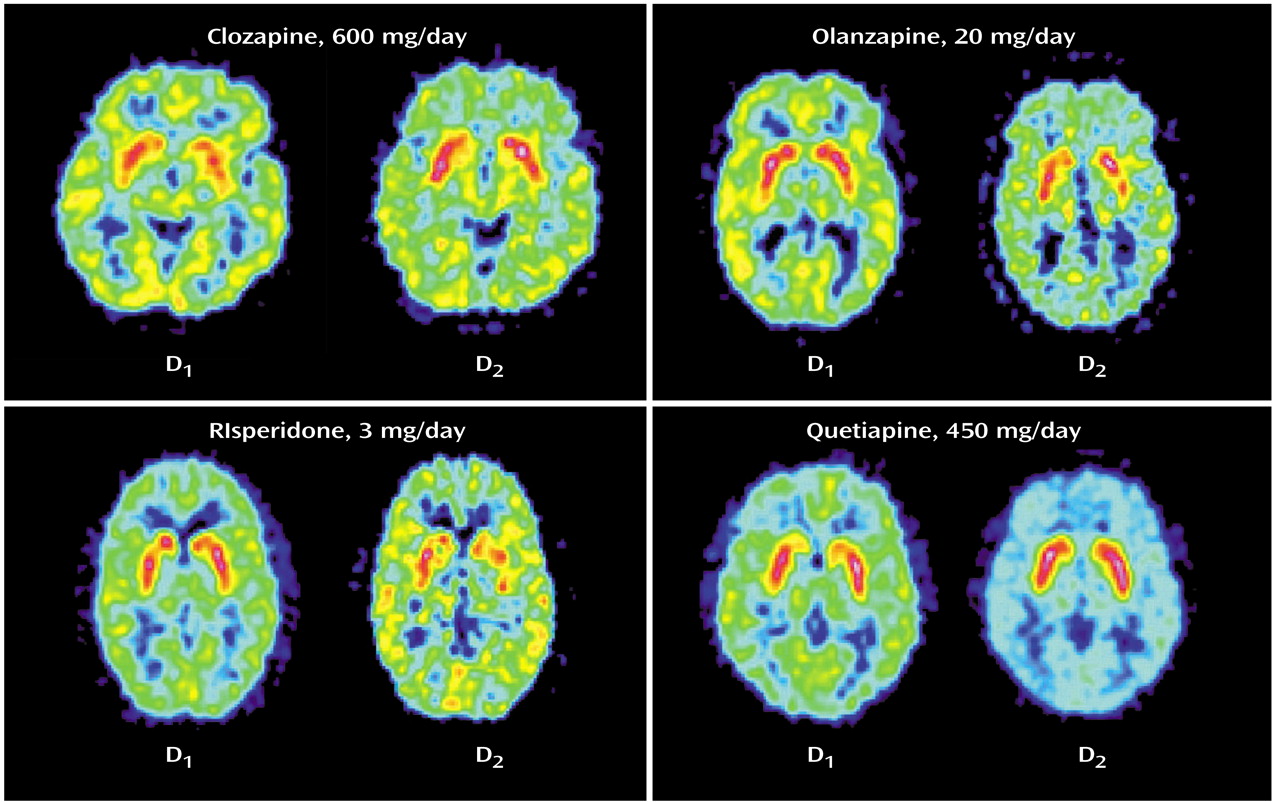
The mean striatal D
1 occupancies ranged from 55% with clozapine to 12% with quetiapine, with the following rank order: clozapine > olanzapine > risperidone > quetiapine (
Table 2). An ANOVA revealed that the differences in striatal D
1 occupancy between groups were statistically significant (F=17.122, df=3, p<0.001). Post hoc Scheffé tests showed that the differences in D
1 occupancies of clozapine versus risperidone (p=0.001) and versus quetiapine (p<0.001) were statistically significant, whereas the difference between clozapine and olanzapine did not reach statistical significance (p<0.36).
In the striatum, the D
2 occupancy ranged from 81% for risperidone to 30% for quetiapine, with the following rank order: risperidone > olanzapine > clozapine > quetiapine. The differences in striatal D
2 occupancies were statistically significant between groups (F=28.770, df=3, p<0.001). Post hoc tests revealed that clozapine showed a significantly lower D
2 occupancy than risperidone (p<0.02) and olanzapine (p<0.04), but significantly higher D
2 occupancy than quetiapine (p=0.001). The ratio of striatal D
1/D
2 occupancy as an index for a “balanced” or equivalent occupancy of D
1 and D
2 receptors was significantly higher for clozapine versus olanzapine (F=23.174, df=1, p=0.001), quetiapine (F=7.365, df=1, p<0.03), or risperidone (F=87.736, df=1, p<0.001) (
Figure 1,
Table 2).
Mean prolactin plasma levels were significantly different among the groups (F=8.421, df=3, p=0.001) and ranged from 9.0 μg/liter with clozapine to 15.3 μg/liter with olanzapine and quetiapine, and 39.2 μg/liter with risperidone. Post hoc tests revealed that plasma prolactin levels with risperidone were significantly higher than with any of the other three antipsychotics (p=0.001). However, there was no statistically significant difference between clozapine, olanzapine, and quetiapine. Furthermore, there was no significant correlation between plasma prolactin levels and striatal D1 (r=–0.36, df=23, p<0.09) or striatal D2 (r=0.30, df=23, p<0.20) occupancy.
Discussion
Clozapine occupies dopamine D
1 and D
2 receptors in vivo, which is in line with previous studies that used PET and [
11C]SCH23390
(23) or [
11C]raclopride
(11). Our study represents the first attempt to compare clozapine’s action on D
1 and D
2 receptors to that of other novel antipsychotics—and we found that it is unique in this regard.
Our receptor occupancy results correspond with the D
1 and D
2 receptor affinity values found in preclinical studies: the K
i values for clozapine were 290–540 nM for D
1 and 130–150 nM for D
2, suggesting relatively similar affinity for both receptors
(22,
33). Corresponding to the comparatively higher D
2 than D
1 occupancy with quetiapine and risperidone in our in vivo PET study, preclinical data suggest a 10- to 100-fold higher affinity for D
2 receptors than for D
1 receptors for both quetiapine and risperidone
(22,
33). We found that olanzapine came closest to clozapine with regard to the “balanced” or equivalent occupancy of D
1 and D
2 receptors, still showing a D
1/D
2 occupancy ratio of 0.54. In line with that, preclinical data showed olanzapine’s K
i values at D
1 of 52 nM and at D
2 of 20 nM
(33).
While we observed occupancy of D
1 receptors, there is no broad consensus on clozapine’s intrinsic efficacy at these receptors. In a number of in vivo assays, clozapine has some preferential, although not selective, action to antagonize D
1 receptor-mediated function
(34). However, D
1 antagonism by itself has not been an effective antipsychotic principle: studies with the selective D
1 antagonists SCH23390
(35), SCH39166
(36–
38), and NNC 01–0687
(39) by themselves were ineffective as antipsychotics. In addition, relatively brief treatment with SCH39166 in doses inducing a more than 70% occupancy of striatal D
1 receptors failed to induce antipsychotic action
(40).
On the other hand, there is some evidence that clozapine behaves as a D
1 agonist: hypothermia produced by clozapine in rats was fully antagonized by either of the selective D
1 receptor antagonists SCH23390 or NNC 01–687
(41). This aspect could be interesting given the clinical and laboratory observations implicating D
1 receptor agonism in the prefrontal cortex in cognitive functions
(41,
42). Finally, regardless of its agonist/antagonist action, a recent [
18F]fluorodeoxyglucose PET study in patients suffering from treatment-resistant schizophrenia showed that brain metabolic and clinical responses to clozapine were related to D
1 receptor genotype
(43). After 5 weeks of treatment with clozapine, brain metabolic decreases were found in patients with the 2,2 but not the 1,2 D
1 receptor genotype. Moreover, patients with the 2,2 D
1 genotype significantly improved with clozapine, whereas those with a 1,2 D
1 genotype did not
(43).
We did not observe a simple relationship between prolactin plasma levels and D
1 or D
2 occupancy rates. This can be explained by the differential blood-brain disposition of the atypical antipsychotics under investigation. In line with our findings, risperidone has been shown to lead to higher prolactin levels than clozapine, olanzapine, or quetiapine
(44). This fact is not directly related to dopamine receptor occupancy in the brain but is mainly due to differential blood-brain barrier penetration of atypical antipsychotics. It has been shown that risperidone has a comparably higher central to peripheral potency for prolactin elevation than olanzapine
(44). Compounds with a higher peripheral potency bring about higher prolactin levels for a given level of functional central antagonism, and thus one cannot expect a simple linear relationship between plasma prolactin and dopamine receptor occupancy with different antipsychotics.
There are several limitations of the current study that suggest caution in how these results are interpreted. We compared D
1 and D
2 receptor binding potential values of patients treated with clozapine and other atypical antipsychotics to that of healthy subjects, since the patients were already receiving treatment and it is very difficult to find neuroleptic-naive patients with similar demographic characteristics. However, this is unlikely to induce a systematic bias in our results, since there is no clear evidence for alterations of striatal D
1 or D
2 receptor number in schizophrenia
(45–
50). Moreover, the main intent of the study was to compare antipsychotics. Since the same baseline was used for all of the agents, it is unlikely to have given rise to differences among antipsychotics.
Plasma drug levels were positively correlated with striatal D
2 receptor occupancies in clozapine- and olanzapine-treated patients but not with quetiapine or risperidone, nor was there such a correlation between plasma drug levels and D
1 occupancy. This is surprising but may be due to the fact that subjects were not randomly assigned to different doses. The apparent difference in the relationships between drug plasma levels and receptor occupancies on D
1 and D
2 receptors can partly be explained by differences in the central and plasma kinetics of the four antipsychotics. The [
11C]SCH23390 PET scans to determine D
1 occupancy were performed at around peak plasma levels for all antipsychotics, while the PET scan with [
11C]raclopride was performed 3–4 hours later. Different time points of PET scans with regard to intake of the last dose of the medication do not make a difference with antipsychotics that show a sustained high blockade of dopamine receptors, such as olanzapine and risperidone
(51). However, it is conceivable that with clozapine and quetiapine, given their more rapid decline from peak plasma concentration
(26,
52), the second scan may have underestimated the peak occupancy values for D
2 receptors.
In summary, this PET study in schizophrenia patients is consistent with the idea that clozapine has a unique interaction with the D1/D2 system as suggested by animal models. The relatively equivalent D1/D2 occupancy may explain the clinical uniqueness of clozapine in patients with refractory symptoms. These cross-sectional data provide a strong impetus for prospective clinical studies focusing on the role of dopamine D1 receptors, with the caveat that it is still unclear whether agonistic or antagonistic properties are desirable, along with moderate D2 antagonism as a means for enhanced therapeutic efficacy against psychosis.