The enzyme catechol
O-methyltransferase (COMT) is critical in the metabolic degradation of synaptic dopamine and norepinephrine
(1), key neurotransmitters hypothesized to influence human cognitive function
(2). The COMT gene contains a functional polymorphism (Val
158Met) that determines high and low activity of this enzyme
(1). Homozygosity for the low-activity (Met) allele is associated with a three- to fourfold reduction of COMT enzyme activity compared with homozygotes for the high-activity (Val) variant, resulting in reduced degradation of synaptic catecholamines in individuals with the Met allele
(3). Recent evidence suggests that in both healthy volunteers and schizophrenia patients, the Met allele is associated with superior performance on measures of prefrontal cortical function
(2,
4).
The 22q11.2 deletion syndrome (DiGeorge/velocardiofacial syndrome) results from a hemizygous deletion in chromosome 22
(5) and is characterized by dysmorphia, cleft palate, and cardiac anomalies
(6). Patients also display a unique behavioral phenotype involving particular deficits in executive function, attention, and abstraction
(7); visuospatial cognition
(8); and elevated rates of ADHD and psychosis
(9–
11). Because the COMT gene maps to the deleted region, the characteristic behavioral manifestations of this syndrome may be related to dopamine dysregulation resulting from COMT haploinsufficiency
(3). Moreover, it is unknown whether COMT genotype in the intact chromosome in patients with 22q11.2 deletion syndrome has a similar influence on executive cognition to that observed in other populations. While prior research has suggested an association between the Met allele and psychopathology in patients with 22q11.2 deletions
(11), no previous work has investigated the effect of COMT polymorphism on neurocognitive function in this population. Thus, the goal of this research was to assess prefrontal cognition in relation to COMT genotype in patients with the 22q11.2 deletion syndrome.
Method
Participants were recruited through the Clinical Genetics Center at the Children’s Hospital of Philadelphia. Genetic diagnosis was confirmed by fluorescence in situ hybridization with the N25(D2S75) molecular probe. Complete neuropsychological and genotype data were available for 44 patients with 22q11.2 deletion syndrome (27 female, 17 male; mean age=11.1 years [SD=3.2]; Val hemizygous: N=28 [64%], Met hemizygous: N=16 [36%]). Forty-two participants were Caucasian, one was black, and one was Asian. All provided written informed consent/assent.
The neurocognitive battery included measures of general intellectual function, memory, language, attention, executive functions, and visuomotor skills. A complete description of the battery and test results are published in detail elsewhere
(7,
8).
COMT genotype was determined by polymerase chain reaction/restriction fragment length polymorphism analysis, as described previously
(1,
12).
The objective of our statistical analysis was to examine the association of COMT genotype with measures of prefrontal cognition. We selected the following executive function measures from our larger battery on the basis of previous factor analytic studies
(13): verbal category fluency (animal naming), Trails B, WISC-3 Arithmetic, and Digit Span tasks. Variables were rescaled to z score equivalents, and an executive function domain score was computed by averaging the z scores on contributing variables. Two-tailed analysis of covariance (ANCOVA) was used to compare the two genotype groups (Met hemizygous versus Val hemizygous). Secondly, we examined individual executive function tests as predictors of allele status. Because there was a tendency toward higher full-scale IQ in Val-hemizygous patients than in Met-hemizygous patients (mean=77.6 [SD=10.5] versus 71.8 [SD=11.4], respectively; F=2.98, df=1, 42, p=0.09), statistical analyses controlled for full-scale IQ.
Results
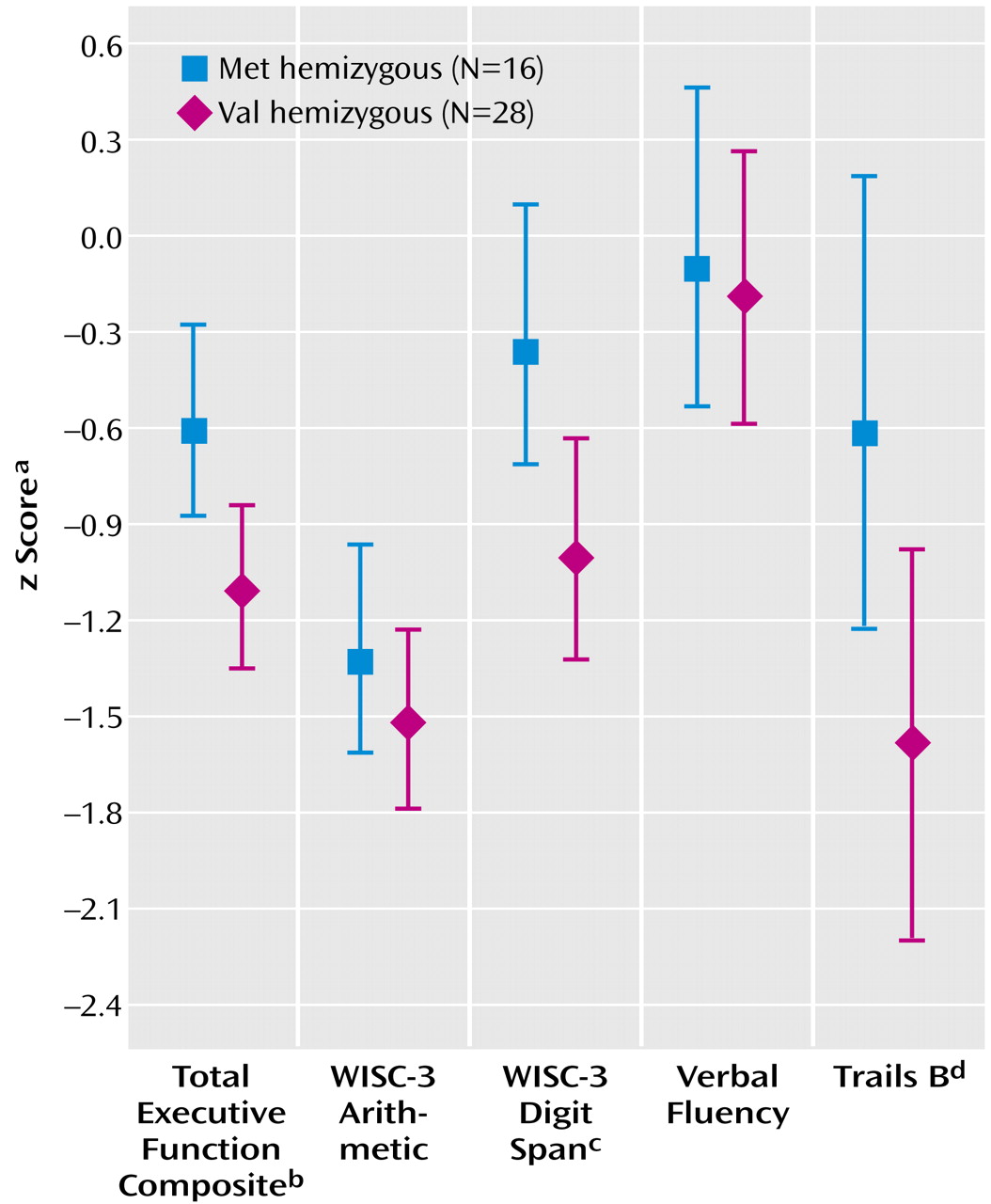
Met-hemizygous and Val-hemizygous subjects were similar with regard to age, sex, and race. In the ANCOVA for executive function composite score, IQ was a highly significant covariate (F=32.48, df=1, 41, p<0.001). Notably, genotype remained a significant predictor of executive function score after effects of IQ were controlled, with Met-hemizygous patients performing better than Val-hemizygous patients (
Figure 1). Met allele was associated with significantly better performance (after IQ was controlled) on the digit span task and superior performance on the Trails B test. Performance on verbal fluency and arithmetic tasks did not differ between groups. There was no main effect of gender nor a genotype-by-gender interaction.
In addition, because the digit span and arithmetic tasks contribute to the full-scale IQ measure, we recalculated our composite using only those tests unrelated to full-scale IQ (Trails B, verbal fluency) and obtained a similar result (F=3.40, df=1, 41, p=0.07).
Discussion
The principal finding of this study is that Met-hemizygous patients performed significantly better than Val-hemizygous patients on a composite measure of executive cognition. Post hoc analyses indicated that this difference was principally driven by performance on the digit span and Trails B tests. This finding appears qualitatively similar to that observed in individuals with no 22q11.2 deletion. However, because our executive function measure involved a broader range of tests than those used in previous studies, which examined the Wisconsin Card Sort as the only dependent measure
(2,
4), it is unclear if COMT genotype explains a greater portion of variance in executive cognition in patients with 22q11.2 deletion syndrome.
In this study group there was a tendency toward higher full-scale IQ in Val-hemizygous patients. Because this relationship was not observed in previous studies of normal subjects
(2,
4), we opted to control for it statistically. However, it is conceivable that this difference is representative of the 22q11.2 deletion syndrome population and thus worth considering in future samples.
To our knowledge, no previous study has investigated cognitive measures in relation to COMT genotype in patients with 22q11.2 deletion syndrome. Investigators have reported increased prevalence of schizophrenia associated with the deletion
(9,
10) and identified schizophrenia susceptibility loci within the 22q11 region
(14), although Murphy et al.
(10) detected no association between COMT genotype and schizophrenia diagnosis in adults with 22q11.2 deletion syndrome. The functional implications of COMT haploinsufficiency are unclear, although patients should be particularly susceptible to the development of psychosis (via increased brain dopamine levels) if the nondeleted chromosome encodes the low-activity (Met) variant of COMT
(3). This is in contrast with the reported association of Val allele load with impaired prefrontal function, and increased risk of schizophrenia
(2). However, the actual effects of 22q11.2 deletion on catecholamine neurotransmission are not known. Future research should examine more direct measures of catecholaminergic turnover in order to quantify prefrontal dopamine uptake as a function of genotype in patients with 22q11.2 deletion syndrome.