Relationship Between High-Risk Haplotype and Clinical Features
The main purpose of this study was to determine whether variation in a gene that has been associated with schizophrenia in several samples imparted risk for a more or less specific form of illness. This question has two important and related dimensions—one theoretical and one practical.
Most important, it will be necessary to understand whether the clearly heterogeneous group of syndromes collectively placed under the rubric of schizophrenia is one or many diseases. The current state of our neurobiological understanding of schizophrenia has not allowed a clear-cut disease process, necessary for the validation of any disease, to be put forward. The greatest level of detail in this understanding yet achieved is the implication of genetic variation in specific genes, such as DTNBP1, in illness susceptibility, although the causative aberrations in these genes are still unknown. An affirmative answer to the question of whether genetic variation in a gene associated with schizophrenia imparts risk for a specific form of the illness would advance the argument that schizophrenia represents a more or less heterogeneous group of diseases, each of which could be associated with more or less specific clinical features. This view would be in contrast to current nosological schemes, which group several syndromes into one illness entity, and it would in turn have important implications for treatment. According to this view, treatment could theoretically be designed to target specific pathophysiological processes and could be individually tailored to cases meeting criteria that are more refined than those currently available.
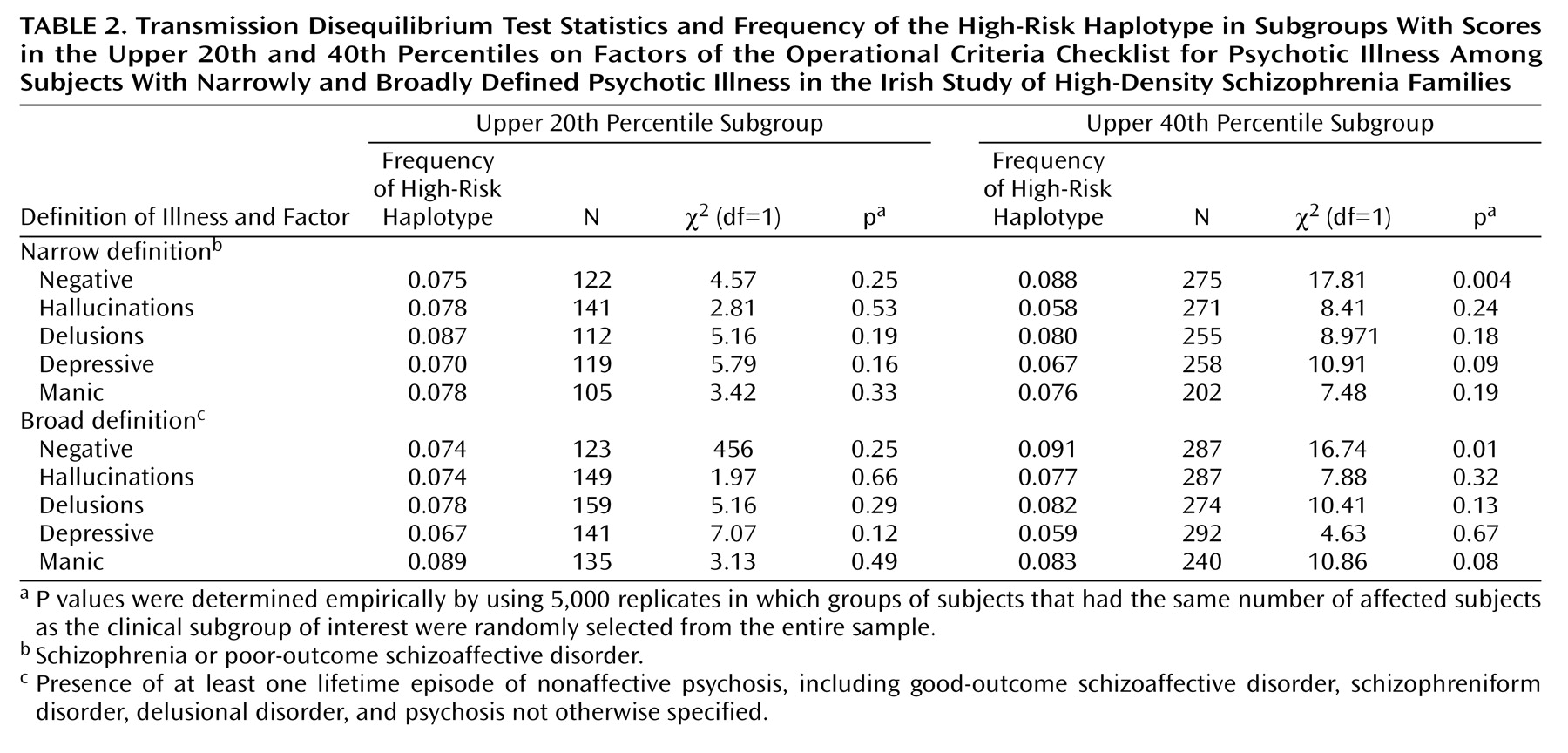
This study provides evidence that genetic variation within a specific gene not only increases the risk of schizophrenia but also does so in a more or less clinically specific manner. The high-risk haplotype was more likely to be transmitted to subjects with high levels of negative symptoms than to other subjects. Several tests were used, and many of the tests were correlated. For example, all Operational Criteria Checklist for Psychotic Illness factors were correlated with at least one other factor, membership in the upper 20th percentile for any factor was a subset of membership in the upper 40th percentile, and the subjects with narrowly defined illness were a subset of the subjects with broadly defined illness. At present, methods of correction for multiple testing in genetic studies have not been universally agreed on. However, it is common to perform linkage analysis by using multiple diagnostic thresholds, as we did in the present study. Although the possibility of false positive results due to multiple testing is acknowledged, strict Bonferroni correction is not often advocated. We therefore interpret our results as highly suggestive and as warranting replication in other samples.
At this stage, the mechanism by which mutations in
DTNBP1 increase liability to schizophrenia is not at all clear, making a potential relationship with negative symptoms even more speculative. Recent neuropathological studies of dysbindin have revealed reduced expression in two areas: the prefrontal cortex
(19) and the hippocampal formation
(20). The two structures are involved in cognitive functions, such as working memory and memory formation, respectively. Although previous factor analyses in this area have extracted a separate cognitive factor
(4), ours did not. However, our negative factor included several items that probably tap cognitive dysfunction. These items included positive and negative formal thought disorder, “incoherent,” “speech difficult to understand,” and “information not credible.” Consistent with this interpretation is the correlation of negative symptoms and cognitive dysfunction found both in patients with first-episode schizophrenia
(35) and in patients with chronic schizophrenia
(36). Furthermore, patients with high levels of negative symptoms perform worse on cognitive tasks subserved by the prefrontal cortex, such as spatial working memory
(37).
Structural and functional aberrations in the prefrontal cortex in particular may be specifically related to the severity of negative symptoms. Subjects with prominent negative symptoms may be more likely to have decreased volume of gray
(38,
39) and white matter
(40) in this region, as well as decreased regional cerebral blood flow to this region
(41,
42). They may also have greater prefrontal neuronal pathology, as evidenced by lower levels of
N-acetylaspartate
(43). More generally, a group of subjects inheriting the high-risk haplotype should be enriched in genetic (and, therefore, biologically predisposed) cases, as phenocopies would not inherit the high-risk haplotype by definition. Patients with prominent negative symptoms may have more severe neurobiological abnormalities than other patients, in addition to those found in the prefrontal cortex. These abnormalities include smaller volume of the temporal cortex
(44–
46), ventricular enlargement
(45,
47,
48), and less cortical folding
(49).
In testing other haplotypes for association with clinical features, an unexpected finding was that haplotype 3 was overtransmitted to subjects in the upper 40th percentile for the depressive symptoms factor. It is noteworthy that this haplotype is composed of the minor allele of p1325 only, which was not significant in single-marker analyses in this sample
(8,
23) and in other samples from Bulgaria
(17) and Ireland
(50). However, it was significant in a German-Israeli sample
(11) and was the only significant individual marker in a Swedish sample
(15). We therefore interpret these results with great caution. Furthermore, because of the lack of any a priori evidence to suggest the association of any other haplotypes with any symptom factor
or with the illness itself, all five additional haplotypes were tested against all five symptom factors, in both diagnostic groups and with both cutoff values. This analysis included a total of 80 additional tests, although again, several Operational Criteria Checklist for Psychotic Illness factors were correlated with each other and with other factors, both within subjects and within sibling pairs. The multiple tests and their exploratory nature make it very difficult to rule out false positive results for the other haplotypes.
Because it will be vital to improve the ability to detect susceptibility genes in the service of their successful identification, it will be important to understand the relationship between the clinical phenotype and genetic linkage and association signals. Several studies have demonstrated that stratification on the basis of clinical features can improve the evidence for linkage at specific chromosomal regions in bipolar disorder
(51), autism
(52), and systemic lupus erythematosus
(53). A recent study by our group demonstrated improved evidence for linkage at a previously linked region when families that were positive for the high-risk haplotype were removed from the analysis (unpublished 2003 data of B. Webb, et al.). These studies imply that making linkage samples as homogeneous for genetic subtypes as possible can reduce the genetic heterogeneity that has been theorized to contribute to the difficulty of studying complex traits. These studies taken together also suggest a relationship between genetic and clinical subtypes. In theory, resources could therefore be used more efficiently if large-scale gene finding studies could maximize the genetic signal-to-noise ratio by targeting clinical, and therefore genetic, subtypes. In breast cancer, use of a sample of early-onset cases increased the evidence for linkage to chromosome 17q
(54), which led to the subsequent cloning of
BRCA1 (55). Substantial effort is currently being dedicated to identifying causative mutations in schizophrenia susceptibility genes and the mechanism by which they increase risk. When such mutations are identified, it will be possible to study the relationship between genetic and clinical heterogeneity more definitively and therefore to better inform psychiatric nosology.
Although sibling pairs were significantly correlated for all factors except hallucinations—suggesting familial influences on the expression of these features—we failed to discern any significant relationships between transmission of the high-risk haplotype and the delusions, manic symptom, and depressive symptom factors. Such familial aggregation could be due to common environmental influences such as prenatal or perinatal events, nutrition, infection, education, social class, and parental factors. Modifier genes, defined as loci influencing clinical features without altering susceptibility, could also be responsible. A genome scan for age at onset
(56) as well as one performed by our group (unpublished 2003 data of A. Fanous, et al.) has supported this suggestion, as has an association study of several candidate genes in our sample
(57). It is also possible, however, that we lacked the statistical power to resolve the true effect of
DTNBP1 variation on these clinical features. Another limitation is that the high-risk haplotype cannot be said to increase risk of illness itself, but it is presumed to be in linkage disequilibrium with one or more causative mutations in
DTNBP1 that have yet to be identified. It is therefore not possible to determine the
degree of correlation of the high-risk haplotype with these mutations, although such a correlation is assumed to exist. Therefore, associations between the high-risk haplotype and clinical features are at best inexact approximations of the relationship between the actual causative variations and clinical features.