Atypical antipsychotic drugs have been widely accepted over traditional antipsychotics for the treatment of schizophrenia. There is, however, lack of agreement regarding differential efficacy among individual atypicals. Some investigators have suggested that the currently available antipsychotics, with the exception of clozapine, have comparable efficacy
(1,
2). Other head-to-head trials have yielded mixed results
(3–
8). Duration of the trial, appropriate dosing, and patient characteristics may account for the inconclusive results. Results from a recent meta-analysis of 124 randomized, controlled trials of 10 atypical antipsychotics versus typical antipsychotics suggest that differences may exist between atypical agents in their ability to reduce psychopathology
(9).
Olanzapine was shown to be superior to placebo or haloperidol on all five factors of the Positive and Negative Syndrome Scale
(10) in a meta-analysis of four large clinical trials (6 weeks in duration) in patients with schizophrenia spectrum disorders
(11). Ziprasidone demonstrated superiority to placebo in a short-term, acute-phase clinical trial
(12) and equivalence to haloperidol
(13,
14) and risperidone
(15) in alleviating psychopathology in patients with schizophrenia. Both ziprasidone
(16) and olanzapine
(17) have demonstrated superiority to placebo in maintaining treatment response in patients with schizophrenia.
It is important to understand the comparative efficacy and safety of atypical antipsychotics for the treatment of schizophrenia, particularly in longer-duration trials. Therefore, we conducted a multicenter randomized, double-blind, parallel-group, 28-week study comparing the efficacy and safety of olanzapine (10–20 mg/day) with those of ziprasidone (80–160 mg/day) in patients with schizophrenia.
Method
Patients
Male or female in- and outpatients (ages 18–75 years) with schizophrenia (DSM-IV) were recruited from 12 sites in Europe and North and South America. The study protocol was approved by local ethical review boards and carried out in accordance with the Declaration of Helsinki. Each patient, or the authorized legal representative, signed an informed consent document that fully explained the risks and benefits of study participation. To be included, the patient had to have a baseline score of 42 or higher on the Brief Psychiatric Rating Scale
(21) and a score of 4 or higher on at least one positive symptom item of the Positive and Negative Syndrome Scale. The patient was also required to have a score of 4 or higher on the severity of illness scale of the Clinical Global Impression (CGI)
(21, pp. 218–222).
Study Design
This was a randomized, double-blind, parallel-group study (trial HGHJ) that was conducted at multiple study sites and lasted 28 weeks. Study period 1 (2–9 days) was the screening, washout, and single-blind placebo lead-in period. Visit 1 consisted of screening tests, medical history, psychiatric and physical examinations, laboratory tests, and two ECGs. Patients were excluded if they had participated in a clinical trial of another drug within 1 month before study entry, had been treated with an injectable depot antipsychotic within one treatment cycle before study entry, or had been treated with clozapine within the 7 days before enrollment. Patients who had used olanzapine or ziprasidone within 6 months of the study’s start and whose treatment had been withdrawn because of clinically important and/or intolerable adverse events or who exhibited a lack of treatment response were also excluded. Patients found to have congenital long QT syndrome, a QTc interval longer than 500 msec, or any ECG abnormality that would confound measurement of the QT interval at visit 1 or 2 were also excluded. Patients were also excluded if they had DSM-IV substance dependence within the past month or had a serious, unstable illness.
Study period 2 was the 28-week double-blind therapy period. Qualifying patients were randomly assigned at a 1:1 ratio, and dosing was determined according to the package inserts: olanzapine at a dose of 10 mg once daily and ziprasidone at 20 mg twice per day, beginning at visit 2. After 3 days, the ziprasidone dose was increased to 40 mg twice per day while the olanzapine dose remained stable. Thereafter, the dose could be increased by one increment at each visit: olanzapine, 5 mg/day to a maximum of 20 mg/day; ziprasidone, 40 mg/day to a maximum of 160 mg/day. The dose could be reduced by the same increment; however, patients were discontinued if they could not tolerate the minimum dose (olanzapine, 10 mg/day; ziprasidone, 80 mg/day). Patients were assessed weekly for the first 2 months and seven additional times thereafter.
Patients who intentionally missed all antipsychotic doses for 5 consecutive days or who took less than 50% of the prescribed doses within one visit interval were removed from the study. Moreover, patients intentionally taking more than the prescribed amount of medication on a regular basis were discontinued.
Lorazepam (≤4 mg/day) was permitted during study period 1. Benzodiazepine or hypnotic monotherapy was permitted during study period 2 (≤10 mg/day of diazepam equivalents recommended). Patients requiring more than two concurrent benzodiazepine hypnotic medications were removed from the study. Benztropine mesylate or biperiden was allowed up to 6 mg/day if extrapyramidal symptoms occurred or existed at visit 1. Once these symptoms had resolved, the dose was reduced as much as possible. Prophylactic use of anticholinergic medication for extrapyramidal symptoms was prohibited.
Assessments
The primary efficacy assessment was reduction from baseline to 28 weeks in the total score on the Positive and Negative Syndrome Scale
(10), each item of which is scored on a scale of 1–7. The secondary efficacy measurements included the Positive and Negative Syndrome Scale subscales evaluating positive and negative symptoms, general psychopathology, cognition, and excitability. Response was defined in the protocol as a 30% or greater improvement in the Positive and Negative Syndrome Scale total score at week 8.
The Positive and Negative Syndrome Scale and the CGI severity of illness scale (22, pp. 218–222) were used to measure symptom exacerbation and time to exacerbation. The Montgomery-Åsberg Depression Rating Scale
(23) and Hamilton Anxiety Rating Scale (22, pp. 194–198) were used to evaluate improvement in depressive symptoms and anxiety, respectively. The CGI improvement scale (22, pp. 218–222) was used to evaluate overall symptom improvement. The Heinrichs-Carpenter Quality of Life Scale
(24) evaluated health-related quality of life.
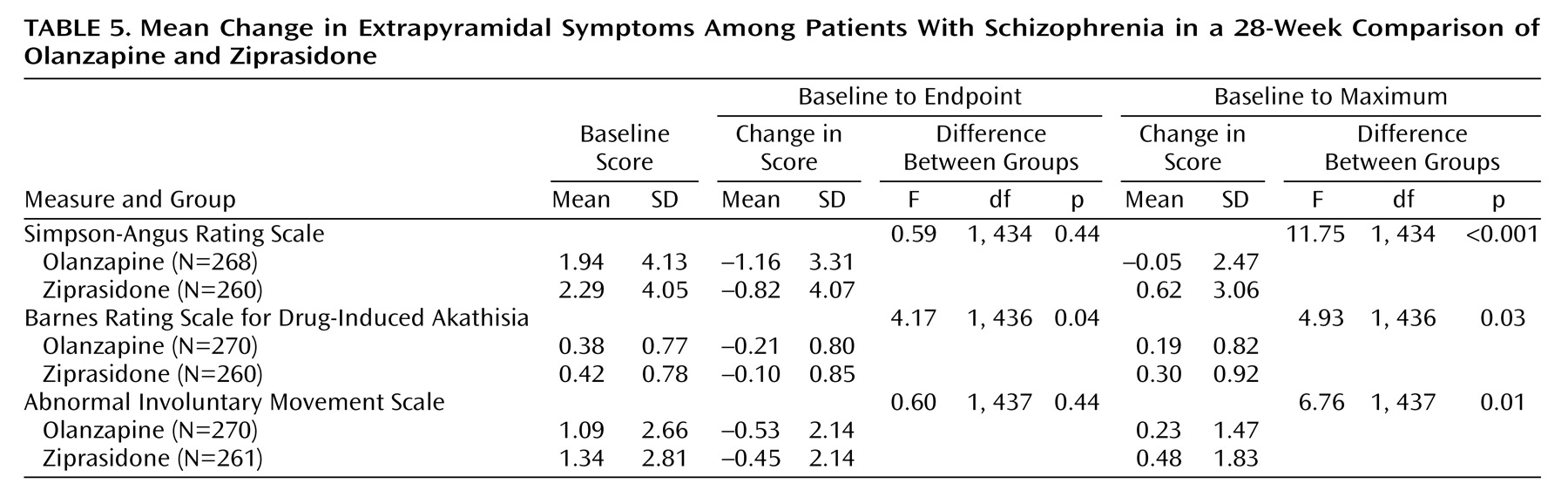
The safety of olanzapine versus ziprasidone was evaluated by comparing rates of treatment-emergent adverse events, vital signs, weight, and results of ECGs and laboratory tests. Fasting levels of glucose and lipids were measured at baseline and weeks 8, 16, 24, and 28. The incidence and severity of extrapyramidal symptoms were measured by the Simpson-Angus Rating Scale
(25), Barnes Rating Scale for Drug-Induced Akathisia
(26), and Abnormal Involuntary Movement Scale (AIMS) (22, pp. 534–537). Both baseline-to-endpoint and baseline-to-maximum changes in scores were assessed.
Statistical Methods
The analyses were done on an intent-to-treat basis unless otherwise specified. Categorical data were analyzed by using Fisher’s exact test. The measures of comparative efficacy and safety—Positive and Negative Syndrome Scale total (primary outcome measure) and subfactors, CGI, depression and anxiety scales, quality of life scale, weight, fasting glucose level, lipid levels, prolactin level, and QTc interval—were determined by using two analytic approaches: mixed-effects model with repeated measures and last observation carried forward. Postbaseline data, i.e., the scores at each visit, were analyzed by using a mixed-effects model with repeated measures (primary data analysis) that included terms for treatment, investigator, visit, treatment-by-visit interaction, baseline value, and baseline-by-visit interaction. Change from baseline to the last observation carried forward was analyzed by using a fixed-effects analysis of covariance including terms for treatment, investigator, and baseline value in the model. Analyses of CGI improvement scores did not include baseline terms, because a baseline CGI improvement score was not determined. All tests of hypotheses were considered significant if the two-sided p value was less than 0.05. Measures of extrapyramidal symptoms were analyzed in two ways as specified in the protocol: mean change to endpoint and change to maximum. SAS version 8.2 (SAS Institute, Cary, N.C.) was used for the statistical analysis.
Results
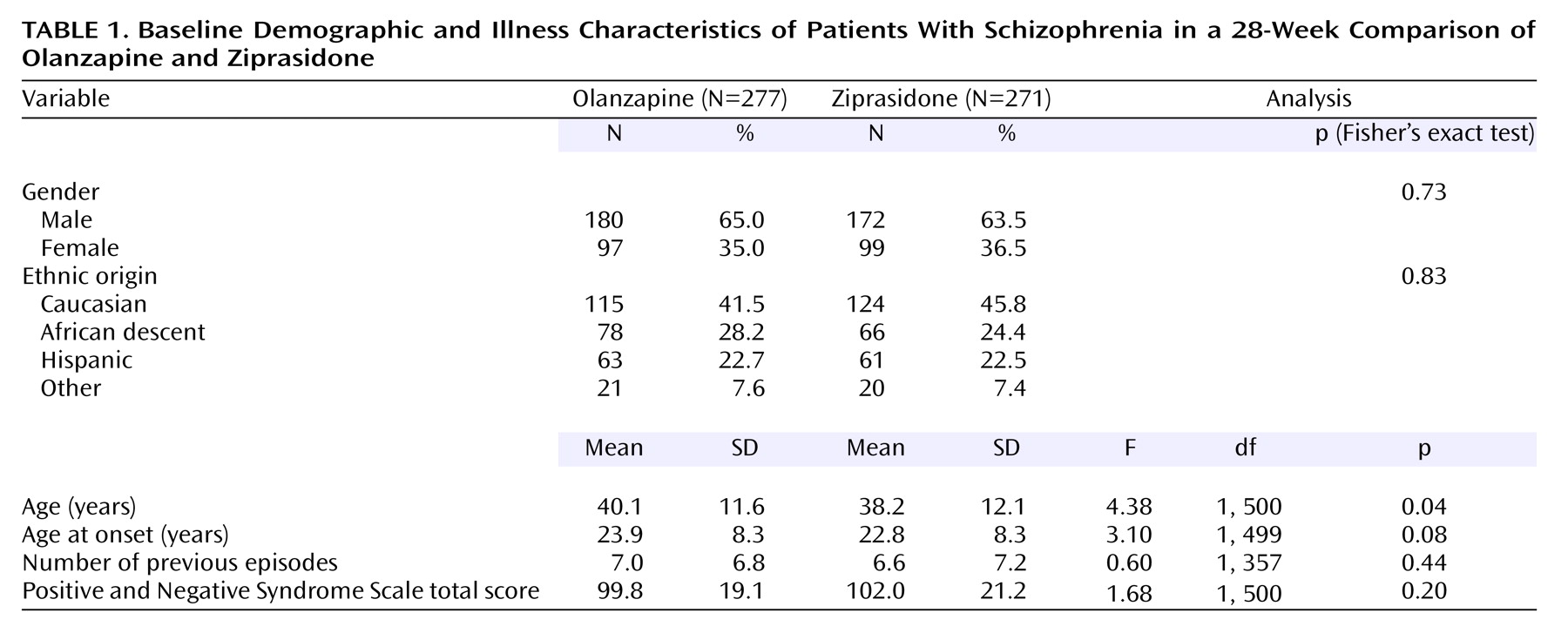
Patient Baseline Demographic and Illness Characteristics
A total of 548 patients were randomly assigned to olanzapine (N=277) and ziprasidone (N=271) (
Table 1). Age was the only baseline value for which there was a statistically significant difference between the two groups. The majority of patients were male (64.2%), and the primary ethnic groups were Caucasian (43.6%), African descent (26.3%), and Hispanic (22.6%).
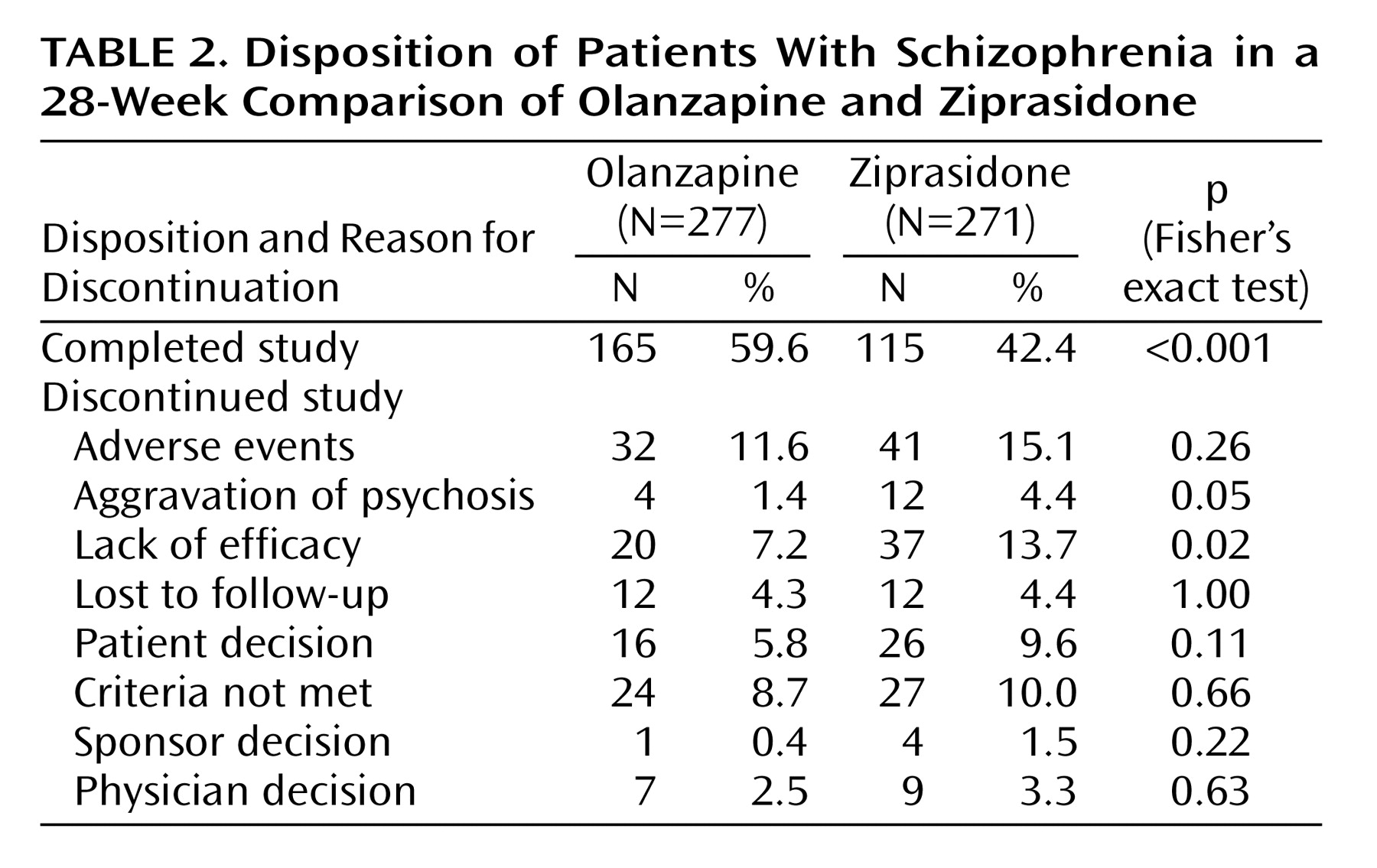
Patient Disposition
Significantly more olanzapine-treated patients than ziprasidone-treated patients completed the study (
Table 2). Among the reasons for discontinuation, only lack of efficacy and aggravation of psychosis were significantly different between groups (both differences favored olanzapine).
Treatment Characteristics
The mean modal doses were 15.27 mg/day (SD=4.52) for olanzapine and 115.96 mg/day (SD=39.91) for ziprasidone. Dose escalation was similar in the two groups, with 83.5% of olanzapine-treated and 92.5% of ziprasidone-treated patients reaching their modal dose by week 4. The change in mean daily dose by week showed similar trends in the two groups. The mean proportions of patients compliant with the study drug regimen were statistically different, 97.8% (SD=0.1) for olanzapine and 94.9% (SD=0.1) for ziprasidone (F=15.04, df=1, 492, p<0.001). Analysis of dose by inpatient or outpatient status at study entry showed that for both drugs the mean modal dose was slightly higher for inpatients than for outpatients (olanzapine: mean=16.74 mg, SD=3.76, N=43, versus mean=15.00 mg, SD=4.6, N=230; ziprasidone: mean=123.08 mg, SD=42.44, N=39, versus mean=114.74 mg, SD=39.43, N=228). However, this difference was statistically significant only in the olanzapine group (F=5.20, df=1, 271, p=0.02). The total score on the Positive and Negative Syndrome Scale at entry did not significantly differ between inpatient and outpatient groups treated with either drug.
The percentage of patients taking at least one dose of a benzodiazepine was significantly greater in the ziprasidone group than in the olanzapine group (53.5% versus 40.4%) (p=0.003, Fisher’s exact test). In addition, significantly more ziprasidone-treated patients took a benzodiazepine for 1–14 days (22.9% versus 14.8%) (p=0.02, Fisher’s exact test) but not for durations exceeding 14 days (30.6% versus 25.6%) (p=0.22, Fisher’s exact test). Significantly more ziprasidone-treated patients than olanzapine-treated patients received at least one dose of an anticholinergic (15.5% versus 7.2%) (p=0.003, Fisher’s exact test). In addition, significantly more ziprasidone-treated patients took an anticholinergic for 1–14 days (8.9% versus 1.4%) (p<0.001, Fisher’s exact test) but not for durations of more than 14 days (6.6% versus 5.8%) (p=0.73, Fisher’s exact test).
Efficacy
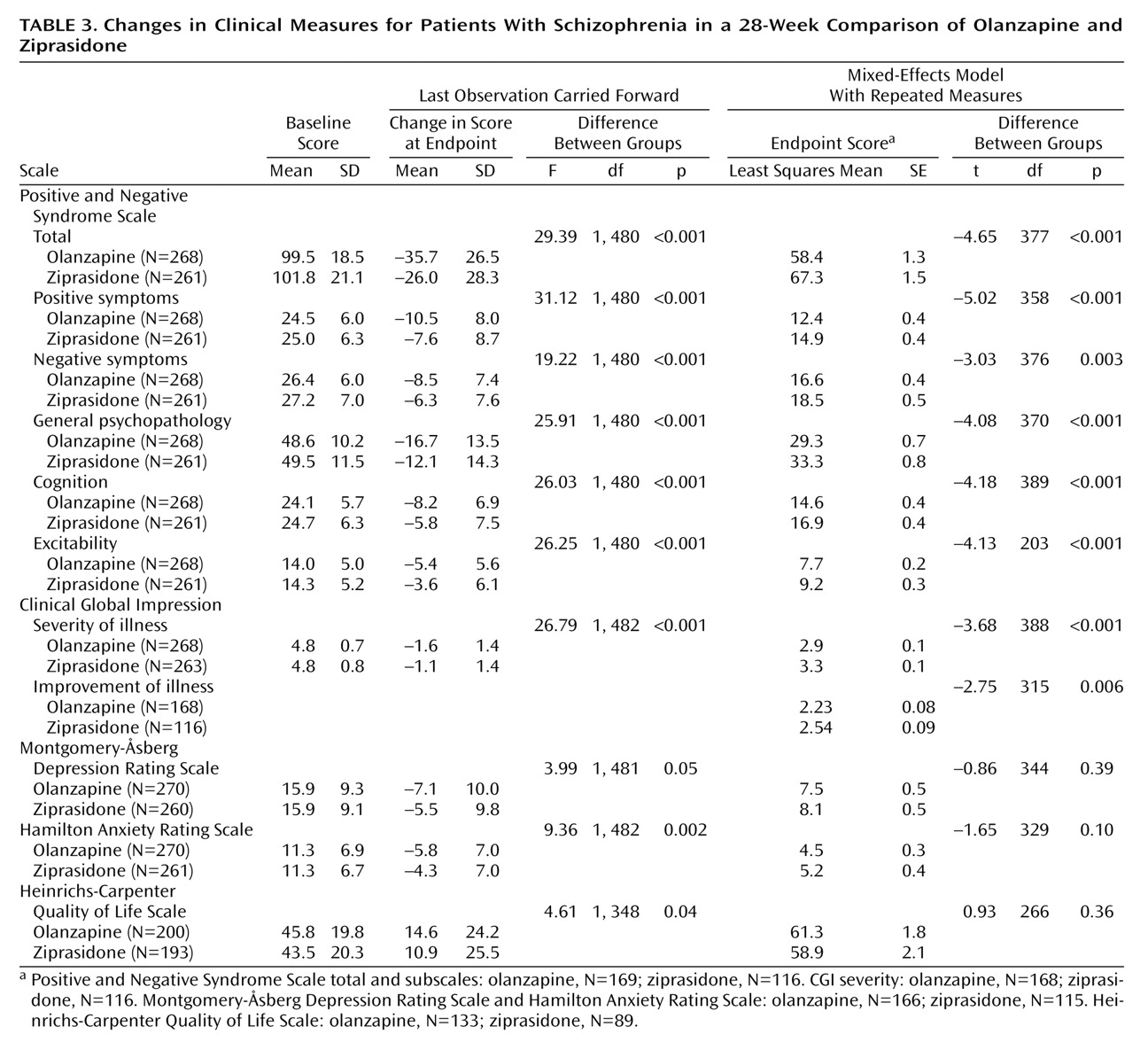
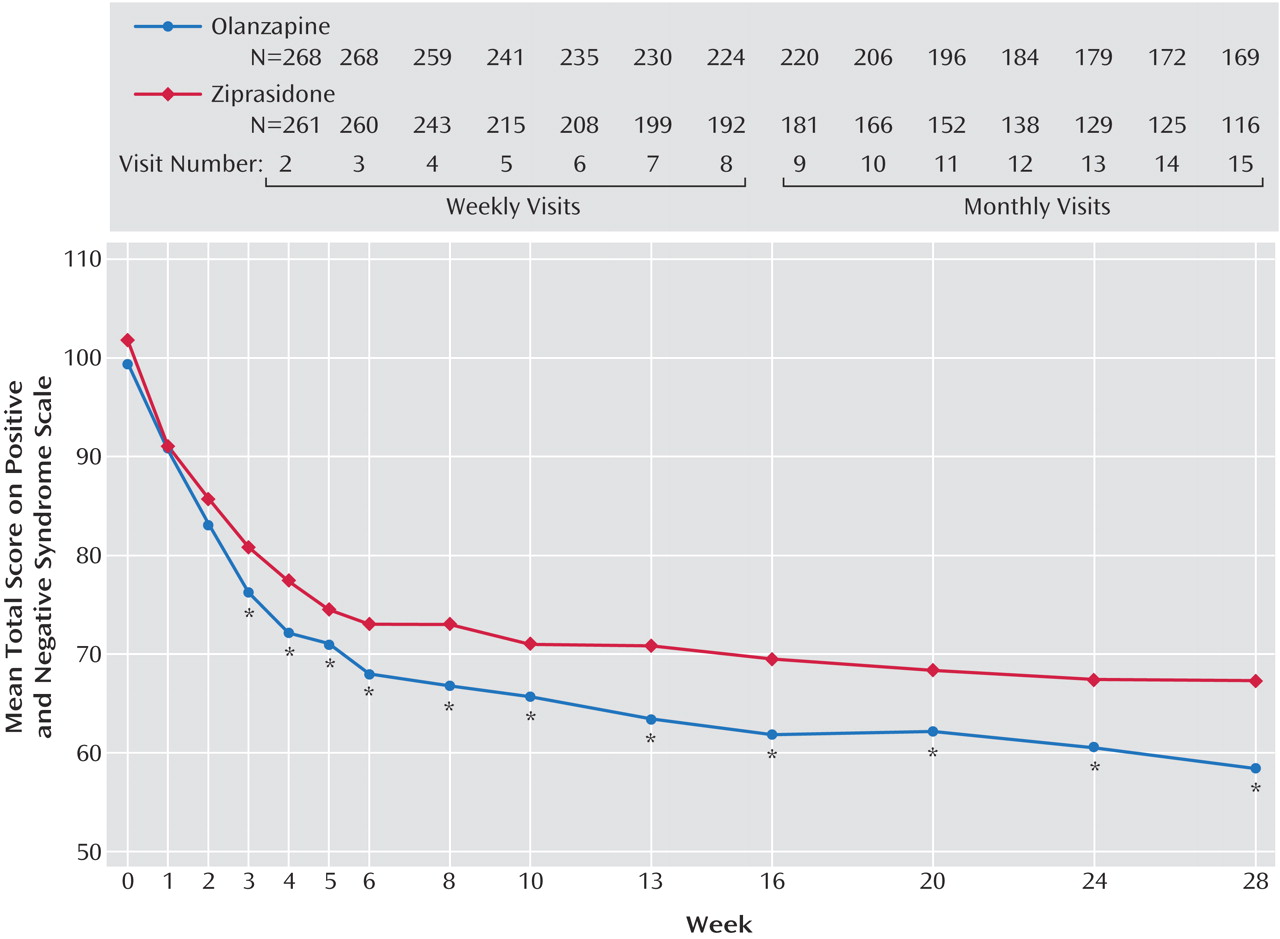
On the primary efficacy measure, the Positive and Negative Syndrome Scale total score, the olanzapine-treated patients showed significantly greater improvement than the ziprasidone-treated patients, according to both the mixed-effects model with repeated measures and analyses with the last observation carried forward (
Table 3). The difference appeared as early as week 3 and was present at all time points from week 6 to endpoint (
Figure 1). The olanzapine-treated patients demonstrated significantly greater baseline-to-endpoint improvement than the ziprasidone-treated patients (
Table 3). The olanzapine-treated patients also showed superior baseline-to-endpoint improvement on the Positive and Negative Syndrome Scale subscales compared with the ziprasidone-treated patients (
Table 3). Separation began at week 2 or 3 and was sustained out to 28 weeks for the subscales for positive symptoms, negative symptoms, general psychopathology, cognition, and excitability.
The CGI severity and improvement scores also showed greater improvement in the olanzapine-treated patients than in the ziprasidone-treated patients (
Table 3). The olanzapine-treated patients showed significantly more improvement (p<0.05) than the ziprasidone-treated patients at all visits from week 3 through endpoint.
The olanzapine-treated patients showed significantly greater baseline-to-endpoint reductions (last observation carried forward) than the ziprasidone-treated patients in the total scores on the Montgomery-Åsberg Depression Rating Scale, Hamilton Anxiety Rating Scale, and Heinrichs-Carpenter Quality of Life Scale (
Table 3). However, there were no significant between-group differences according to the mixed-effects model with repeated measures (
Table 3).
Response Rate and Time to Symptom Exacerbation
Response was defined in the study protocol as a 30% improvement in Positive and Negative Syndrome Scale total score at endpoint, and the rate was significantly higher for the olanzapine group than for the ziprasidone group (58.6% versus 42.5%) (p<0.001, Fisher’s exact test). Exacerbation of symptoms was defined in the study protocol as a worsening of 20% or more in the Positive and Negative Syndrome Scale total score and a worsening in the CGI severity of illness score of 1 point or more after week 8. There was no significant difference in the percentage of patients who experienced exacerbation of symptoms between treatment groups, 14.6% for olanzapine and 25.3% for ziprasidone (p=0.06, Fisher’s exact test).
Safety
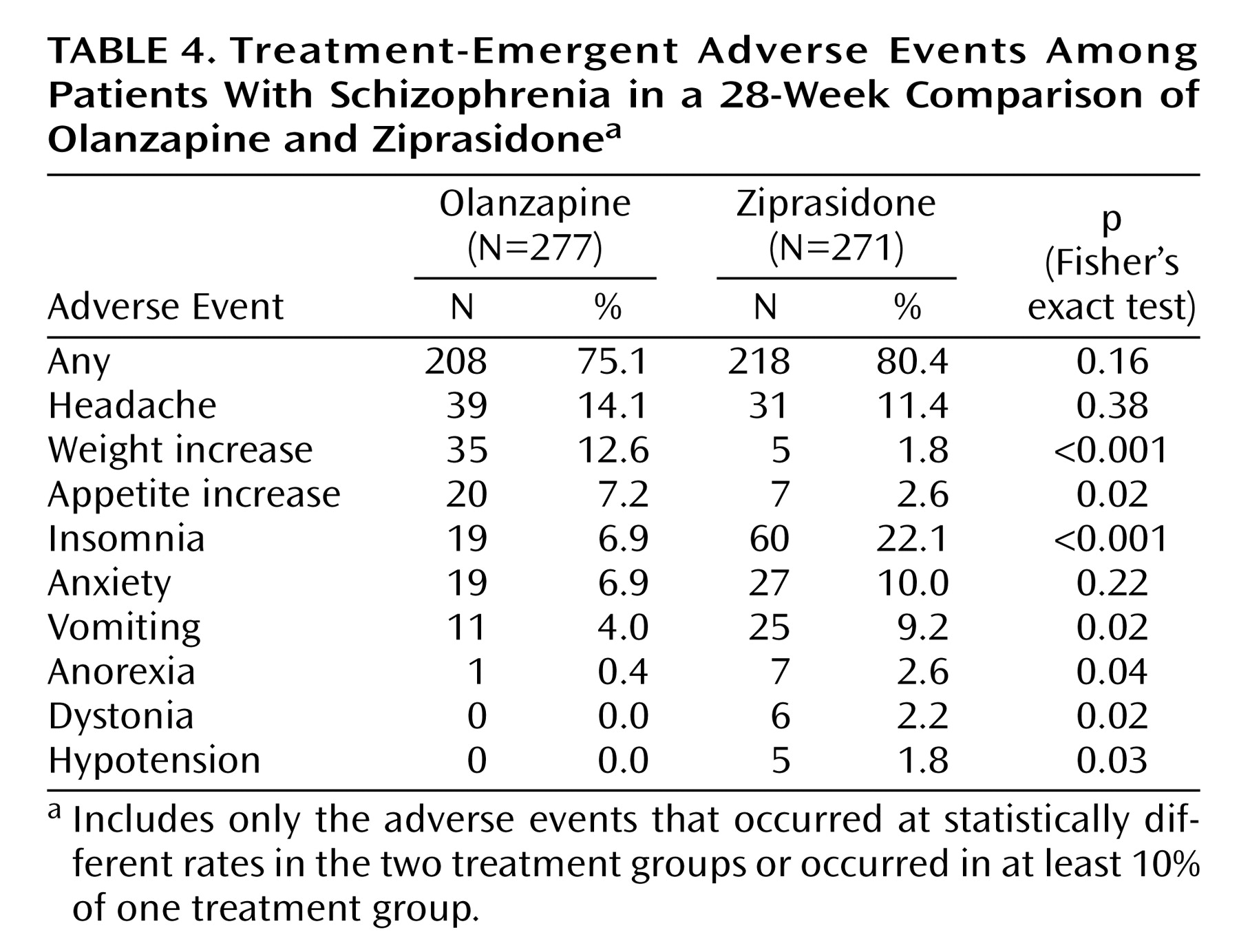
Treatment-emergent adverse events, i.e., events that first occurred or worsened during treatment, were experienced by 75.1% of the olanzapine-treated patients and 80.4% of the ziprasidone-treated patients (
Table 4). The treatment-emergent adverse events experienced by significantly more olanzapine- than ziprasidone-treated patients were weight increase and appetite increase (
Table 4). The adverse events experienced by significantly more ziprasidone- than olanzapine-treated patients were insomnia, vomiting, anorexia, dystonia, and hypotension (
Table 4).
There was no difference between the two treatment groups on mean baseline-to-endpoint changes on the AIMS and Simpson-Angus Rating Scale. There was a significantly greater improvement in mean baseline-to-endpoint change on the Barnes Rating Scale for Drug-Induced Akathisia in the olanzapine treatment group (
Table 5). Analysis of the baseline-to-maximum change showed that the ziprasidone-treated patients had significantly greater maximum changes than the olanzapine-treated patients on the AIMS, Simpson-Angus scale, and Barnes scale (
Table 5).
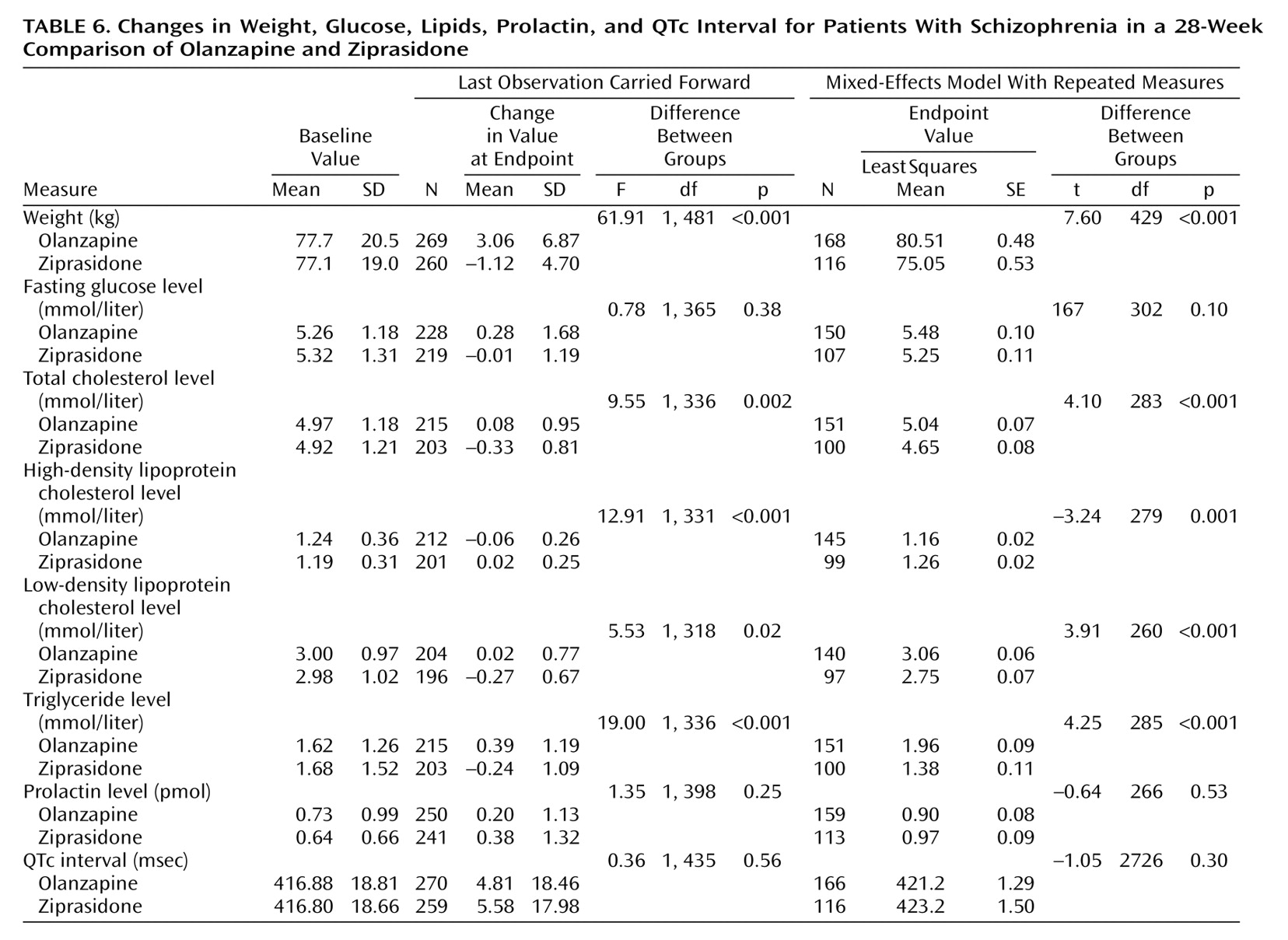
The baseline-to-endpoint changes in fasting glucose and lipid levels, weight, prolactin level, and QTc interval are presented in
Table 6. Using both the analysis of the last observation carried forward and the mixed-effects model with repeated measures, we found significantly greater increases in body weight and levels of total cholesterol, low-density lipoprotein (LDL) cholesterol, and triglycerides and a significantly greater decrease in high-density lipoprotein (HDL) cholesterol in the olanzapine-treated patients than in the ziprasidone-treated patients. There was no statistically significant difference between the groups in the change to endpoint in fasting glucose level, prolactin level, or QTc interval. Insulin levels were not measured.
Discussion
In this study, olanzapine was significantly superior to ziprasidone on the primary efficacy measure of the Positive and Negative Syndrome Scale total score. The difference occurred by week 3 and was maintained throughout nearly the entire 28 weeks. Moreover, significantly greater improvements for the olanzapine group were found on the Positive and Negative Syndrome Scale subscales for positive symptoms, negative symptoms, general psychopathology, cognition, and excitability. For symptom exacerbation (CGI severity) and improvement (CGI improvement), significantly better ratings were seen in the olanzapine group. On the Montgomery-Åsberg Depression Rating Scale, Hamilton Anxiety Rating Scale, and Heinrichs-Carpenter Quality of Life Scale, significantly greater improvement was seen in the olanzapine group according to the analysis with the last observation carried forward but not according to the mixed-effects model with repeated measures. Because there was a higher percentage of dropouts in the ziprasidone group, the analysis with the last observation carried forward may have had a greater likelihood of detecting a statistically significant difference in the case of smaller effect sizes that favor olanzapine.
The differences in efficacy results between this study and the unpublished study by Simpson et al. (a 6-month blinded continuation study of ziprasidone versus olanzapine)
(19) are striking. Whereas olanzapine-treated patients improved significantly more than ziprasidone-treated patients in our study, the improvements were similar in the two treatment groups in the Simpson study (the changes in the total score on the Positive and Negative Syndrome Scale were –33.9 for olanzapine and –32.0 for ziprasidone). In contrast to those in the Simpson study, the olanzapine-treated patients in the present study also had significantly greater improvement in scores on the positive and negative symptom subscales of the Positive and Negative Syndrome Scale. The discrepant results between this study and the Simpson study may be due to drug dosing issues. The Simpson study allowed a maximum olanzapine dose of only 15 mg/day, whereas in this study patients were allowed up to 20 mg/day. The mean daily dose of olanzapine in the present study is similar to those used in previous studies lasting 6 months or longer
(4,
8,
27). The lower dose ceiling in the Simpson study may have prevented the olanzapine-treated patients from showing further improvements in psychopathology. In contrast, one could argue that the lower mean dose for ziprasidone in this study than in the Simpson study could explain the lesser improvement in this study. The dosing followed package insert guidelines. For ziprasidone, the package insert states, “Efficacy in schizophrenia was demonstrated in a dose range of 20 to 100 mg BID in short-term, placebo-controlled clinical trials. There were trends toward dose response within the range of 20 to 80 mg BID, but results were not consistent. An increase to a dose greater than 80 mg BID is not generally recommended”
(28). In this flexible-dosing study, investigators were allowed to increase the ziprasidone dose up to 160 mg/day. Moreover, the mean daily dose of ziprasidone was similar to that in a 44-week continuation study comparing ziprasidone and risperidone
(29) and a 28-week study comparing ziprasidone and haloperidol at a dose of 116.5 mg/day
(14), which showed comparable responses with the two agents. In addition, more than 80% of the patients in both treatment groups were at their modal dose for the study by 4 weeks of treatment, suggesting that differences in dosing schedules would not account for differences in symptom improvement. Flexible dosing has the advantage of allowing investigators to achieve the best balance between efficacy and safety/tolerability and thus mimics clinical practice. However, future studies that include upward forced titration and/or multiple fixed-dose arms would be desirable to address the differential dosing questions raised here. Another issue that may at least partly account for the discrepancy in improvement on the Positive and Negative Syndrome Scale is that in the Simpson study, only early responders (i.e., patients who had a ≥20% improvement in the Positive and Negative Syndrome Scale total score or who had a CGI improvement rating of ≤2) could participate in the 6-month continuation phase.
In the present study, both groups showed continued improvement throughout the entire 28 weeks. In the Simpson study, responders showed a slight worsening in both groups from week 6 through study completion. These findings suggest that many patients might need more than 6 weeks of treatment for maximum benefits. These patients would be considered late responders, and perhaps olanzapine is more effective than ziprasidone in improving psychopathology in these patients, which might explain why olanzapine showed greater efficacy in the present study. Future studies will be needed to test this hypothesis. In the study comparing ziprasidone and risperidone
(15), only patients responding at 8 weeks were allowed to continue for an additional 44 weeks. As in the Simpson study, at the end of the continuation period there were no significant differences in efficacy measures between groups
(15). Thus, to better differentiate the efficacy of the atypical antipsychotics, longer-term studies should be considered, as well as responder extensions or continuation trials.
The discontinuation rate for ziprasidone-treated patients in this study (57.6%) was similar to the rate (55.0%) in the 28-week ziprasidone-versus-haloperidol trial
(14). During the 6-week acute phase of the Simpson trial, 48.5% of the ziprasidone-treated patients discontinued participation, compared to 36.8% of the olanzapine-treated patients
(19). In the patients who continued for 6 months, the discontinuation rates were high (olanzapine, 70.4%; ziprasidone, 69.4%)
(19), with only 3% of the patients in each treatment group discontinuing because of an adverse event. The discontinuation rates for ziprasidone were also high in the risperidone-comparator trial (26.2% in the acute phase and 66.1% in the continuation phase)
(29). In 30-week
(8) and 28-week
(4) studies comparing olanzapine and risperidone for the treatment of schizophrenia, the completion rates were 53.1% and 57.6% for the olanzapine-treated patients, respectively. These completion rates for olanzapine are comparable with the rate in the present study.
There were no significant between-group differences in extrapyramidal symptoms in the change-to-endpoint analyses, but superiority for olanzapine was evident when we examined the change-to-maximum score. This discrepancy may be explained by the fact that more ziprasidone-treated patients required at least one dose of an anticholinergic or anticholinergic therapy for 1–14 days to control extrapyramidal symptoms. In addition, treatment-emergent dystonia occurred in six ziprasidone-treated patients but in no olanzapine-treated patients. In the Simpson study, the olanzapine-treated patients also showed greater improvement of extrapyramidal symptoms, and there was a statistically significant difference in the change on the Barnes Rating Scale for Drug-Induced Akathisia (olanzapine, –0.8; ziprasidone, –0.3) (p≤0.05). In that study, more ziprasidone-treated than olanzapine-treated patients experienced treatment-emergent extrapyramidal symptoms (11.3% versus 4.2%). The Simpson study did not show any data concerning anticholinergic use.
The weight change in both groups in this study was similar to that in previous 6-month studies
(4,
14,
19), with an increase in the olanzapine group and a decrease in the ziprasidone group. As in the Simpson et al. study
(19), there was no significant between-group difference in the baseline-to-endpoint change in fasting level of glucose. Glick et al. reported that the median insulin level and the homeostasis model assessment of insulin resistance (HOMA-IR) were significantly higher after 6 weeks of olanzapine treatment than at baseline; however, only the insulin level was significantly higher in olanzapine-treated patients than in ziprasidone-treated patients
(20). In the study by Simpson et al., after 6 months of therapy, fasting insulin and glucose were significantly higher than at baseline in the olanzapine-treated patients, but they did not significantly differ between the two treatment groups
(19). Because insulin levels were not measured in the present study, it cannot be determined whether similar differences in insulin levels were present and whether there may have been compensatory increases in insulin secretion to maintain normoglycemia within the context of treatment-emergent insulin resistance. The differences in lipid profile changes observed between groups are consistent with the direction of weight change in the groups, as supported by the associations between weight change and mean triglyceride or cholesterol change. The increases in weight, total cholesterol, LDL, and triglycerides and decrease in HDL present long-term risks for coronary heart disease and stroke
(30,
31). It is important for doctors to address these issues through aggressive weight and lipid management or alternative therapy.
In conclusion, olanzapine demonstrated superiority to ziprasidone on the overall Positive and Negative Syndrome Scale, the primary outcome measure, at study endpoint. Significant between-group differences occurred as early as 3 weeks. In addition, greater symptom improvement in olanzapine-treated patients was determined for the Positive and Negative Syndrome Scale positive and negative symptom subscales and the CGI improvement and severity scales. Furthermore, in the analysis using the last observation carried forward, greater improvements in depressive and anxiety symptoms in the olanzapine group were found, as determined with the Montgomery-Åsberg Depression Rating Scale and Hamilton anxiety scale, respectively. The patients treated with olanzapine had significantly greater weight gain and less favorable lipid profiles than the ziprasidone-treated patients. In keeping with the overall risk-versus-benefit assessment that guides physicians in making treatment choices, these findings need to be considered within the overall context of the efficacy and safety profile of olanzapine and ziprasidone.