According to the
N-methyl-
d-aspartate receptor (NMDAR) hypofunction model of schizophrenia
(1,
2), NMDAR blockade results in inhibition of γ-aminobutyric acid neurons, causing disinhibition of glutamatergic neurons that converge onto pyramidal neurons in widely distributed cortical regions. NMDAR hypofunction and/or the downstream surge of glutamate in corticolimbic regions may be related to positive and negative symptoms in schizophrenia. Directly supporting this hypothesis, extracellular glutamate concentrations in the prefrontal cortex were found to increase in awake rats when they were given ketamine, an NMDAR antagonist
(3). However, to our knowledge, no studies assessing the effects of NMDAR antagonism on glutamatergic activity in humans have been reported.
The current study investigated the effects of ketamine on anterior cingulate glutamatergic activity and the relationship to ketamine-induced schizophrenia-like features in healthy humans. Glutamine, the major metabolite of synaptic glutamate
(4,
5), was measured with 4-T
1H proton magnetic resonance spectroscopy (
1H-MRS). We hypothesized that there would be a significant increase in anterior cingulate glutamine with ketamine administration, and that this increase would be directly related to schizophrenia-like positive and negative symptoms.
Method
A double-blind, placebo-controlled, crossover design was applied over two sessions separated by 1–2 weeks. Each session consisted of placebo or ketamine administration, MRS scanning, and behavioral assessments. Subjects were administered ketamine on one day and placebo (saline) on the other in a block-randomized manner.
Ten healthy male subjects completed the study (mean age=24.7 years, SD=3.4). Inclusion/exclusion criteria were 1) no past or present psychiatric disorder as determined with the Structured Clinical Interview for DSM-IV Non-Patient Version
(6), 2) no first-degree relatives with a diagnosis of a psychotic disorder, and 3) no current medical illnesses as determined by a physical examination and laboratory tests. All subjects gave written informed consent and were paid for their participation. This study was approved by the University of New Mexico Institutional Review Board and the U.S. Food and Drug Administration.
Ketamine was administered with a loading dose of 0.27 mg/kg over 10 minutes and a maintenance dose of 0.00225 mg/kg per minute for the remaining extent of the experiment, up to 2 hours
(7). This dose is well below that used for anesthesia and has proven reliable in producing mild schizophrenia-like symptoms with an excellent safety profile
(7).
We used spectroscopic acquisition and analysis methods developed at the University of Western Ontario that have been described in detail elsewhere (8–10). Briefly, bilateral anterior cingulate spectra were acquired from an 8-cc voxel by using
1H-stimulated echo acquisition mode (TR=2000 msec, TE=20 msec, TM=30 msec, dwell time=500 μsec, 256 water-suppressed and 16 water-unsuppressed averages) with a 4-T scanner (Varian, Palo Alto, Calif.). Water suppression was achieved by using three chemical-shift-selective pulses. Spectra were analyzed by using curve-fitting software and normalized to water-yielding quantification of glutamine, as well as the metabolites
N-acetylaspartate, choline, creatine, and glutamate
(8). Three spectra were acquired, the first before ketamine or placebo administration, the second during loading, and the third at the beginning of administration of maintenance doses. Each spectral acquisition took about 10 minutes.
Ratings were conducted by one psychiatrist (J.R.B.) using the Brief Rating Psychiatric Rating Scale (BPRS)
(11), the Scale for the Assessment of Negative Symptoms (SANS)
(12), and the Clinician-Administered Dissociative States Scale
(13) during the drug maintenance phase, approximately 45 minutes after drug start and following the MRS scan. Performance on the Stroop
(14), a task demonstrated to involve the anterior cingulate
(15), was assessed following the behavioral ratings.
Spectroscopic measures were compared between baseline and postinfusion time points with paired t tests separately for each condition (placebo or ketamine). Behavioral measures were analyzed between conditions (placebo versus ketamine) with paired t tests. The relationships between the change in glutamine and behavioral measures were assessed with Pearson product-moment correlations. One-tailed tests were used for glutamine and behavioral measures because of the directional hypotheses, whereas Bonferroni-corrected two-tailed tests (alpha=0.0125) were used for the other four metabolites.
Results
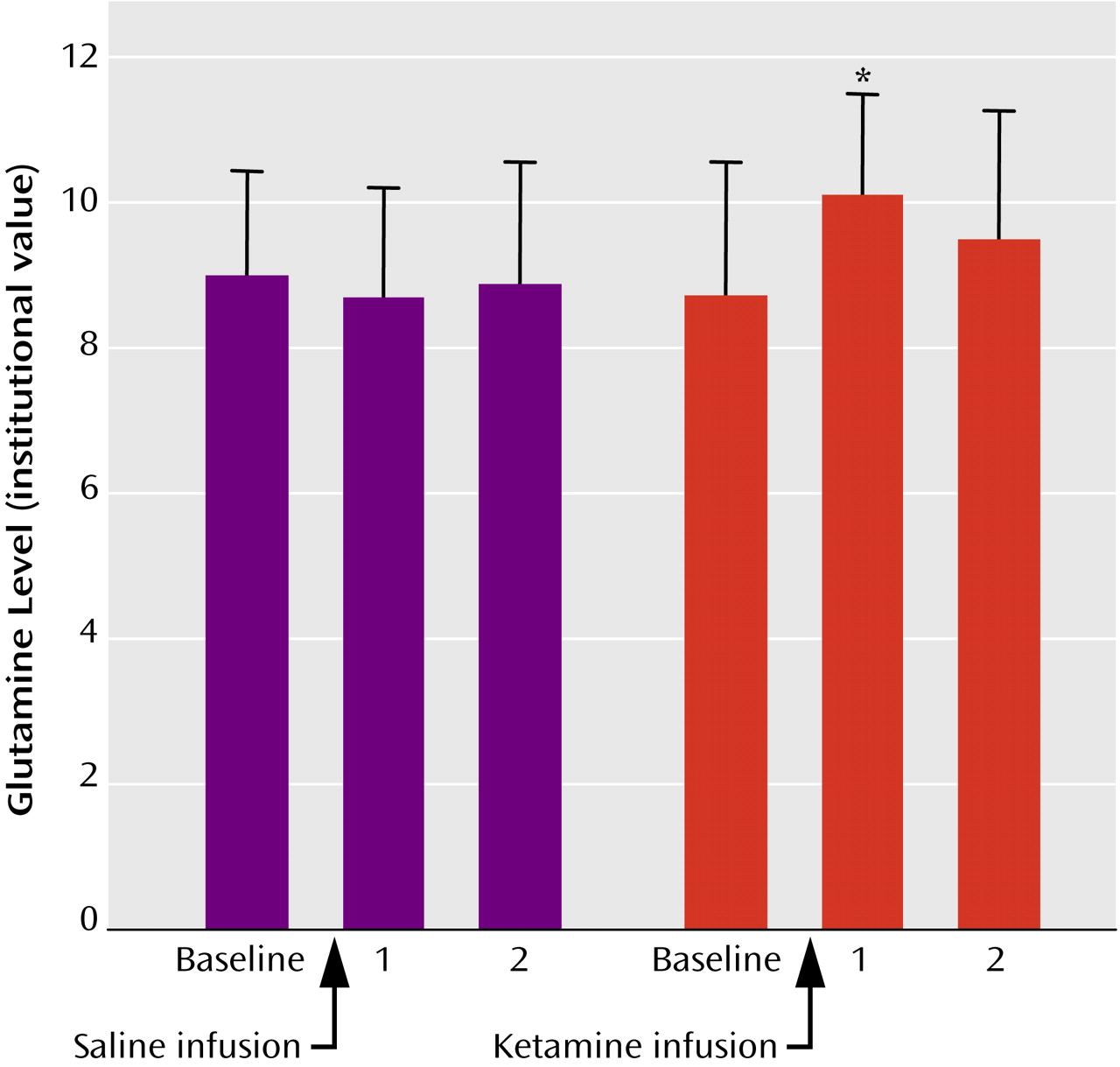
One subject had poor quality spectra and was excluded from the spectral analyses. Another subject did not complete the Stroop task because of nausea associated with ketamine and was excluded from analyses involving Stroop performance. As predicted, results revealed a significant increase in glutamine from baseline to the first time point following ketamine infusion (t=2.0, df=8, p<0.05). The glutamine concentration for the second time point was still elevated when compared with baseline, but not to a statistically significant degree (p>0.05) (
Figure 1). Also, there were no statistically significant differences among other metabolites for baseline and first and second time points following ketamine infusion (
N-acetylaspartate: p=0.36, p=0.18; choline: p=0.33, p=0.08; creatine: p=0.14, p=0.06; glutamate: p=0.17, p=0.55).
Participants experienced schizophrenia-like features associated with ketamine as exhibited by an increase in behavioral rating scores (SANS: t=2.5, df=9, p<0.05; Clinician-Administered Dissociative States Scale: t=4.6, df=9, p<0.001; BPRS: t=1.7, df=9, p=0.06), but, contrary to expectations, these were not significantly correlated with glutamine changes (all p>0.2). However, there was a trend for a negative correlation (r=–0.6, df=7, p=0.06) for Stroop performance and change in glutamine, indicating that increases in glutamine associated with ketamine were related to poorer Stroop performance. However, Stroop scores were not significantly worse during the ketamine condition (mean=5.8, SD=9.5) than the placebo condition (mean=4.3, SD=7.1) (p=0.6).
Discussion
Consistent with our hypothesis, we found a significant increase in anterior cingulate glutamine, a putative marker of glutamate neurotransmitter release, with ketamine administration. Glutamine levels were greatest at the first time point, probably reflecting higher ketamine levels associated with the loading dose. However, mean glutamine levels were still elevated at the second time point, while the maintenance dose was being administered. Providing some indirect evidence for this model, positron emission tomography studies in healthy humans have shown elevations of blood flow
(16) and glucose metabolism
(17,
18) in the anterior cingulate with ketamine, and these elevations were positively correlated with ketamine-induced schizophrenia-like symptoms. In this study, however, glutamine increases were not significantly correlated to schizophrenia-like symptoms induced with ketamine. These inconsistencies may be due to different rating scales
(17,
18), limited variance in the induction of some symptoms with ketamine, or to the possibility that symptoms correlate with a relatively smaller region of the anterior cingulate
(16) than the spectroscopic voxel we studied.
This study revealed glutamine increases to be marginally related to performance on a cognitive test known to involve the anterior cingulate. Therefore, increased anterior cingulate glutamatergic activity may not be related to psychotic symptoms and could be an important catalyst in the deteriorating course (cognitive and social functioning) of schizophrenia. Our finding is consistent with
1H-MRS studies that have shown increased glutamine levels in antipsychotic-naive patients with schizophrenia but no significant relationship to symptoms
(9,
19).
There are several limitations to this study. First, the number of subjects is small. Data must be acquired on more subjects to substantiate the preliminary finding of increased glutamine with ketamine administration and to examine further the relationship between psychotic symptoms and glutamine changes. We were not able to measure glutamate release directly because the total glutamate concentration measured with
1H-MRS is devoted to both metabolism and neurotransmission
(5). Therefore, we measured glutamine, which has been shown to be a good index of the turnover of the synaptic glutamate involved in neurotransmission
(4,
5). Additional regions need to be assessed because it is possible that glutamine increases may be related to psychotic symptoms in different brain areas. Finally, repeated or chronic ketamine administration in humans might be a more valid model of schizophrenia but is not feasible for ethical reasons. Studies with nonhuman animals may be required to document glutamatergic alterations associated with chronic ketamine administration.
To our knowledge, this is the first study in humans to document that NMDAR antagonism results in increased glutamate release in the anterior cingulate. This provides important evidence for a missing component of the NMDAR hypofunction model of schizophrenia. Finally, ketamine challenge with high-field 1H-MRS in normal subjects may provide a paradigm to test pharmaceutical interventions that modulate glutamate, testing their potential use to treat schizophrenia.