In this article, we focus on four psychiatric disorders—depression/mood disorders, posttraumatic stress disorder (PTSD), attention deficit hyperactivity disorder (ADHD), and schizophrenia—because research concerning the neurobiological and mechanistic connections between these disorders and substance use disorders is particularly active. With the rapid development of technical advances in the neurosciences, the amount of information concerning the molecular biology, neurotransmitter systems, and neural circuitry involved in mental illness and substance use disorders has increased dramatically. In this article, we conceptualize chronic distress as a central construct underlying the association of each of these four psychiatric disorders with substance use disorders and examine emerging neurobiological findings within this framework.
Etiological Relationships: Theoretical Perspective
Although convincing data support a strong association between a variety of psychiatric disorders and substance use disorders, the nature of the relationship is complex and may vary depending on the disorder in question and substance that is used. Several theories have been proposed to explain the high co-occurrence. Certain psychiatric disorders may be risk factors for development of substance use disorders or may modify the course of substance use disorders. One of the more overarching theories of addiction is that drugs and their specific psychotropic effects are used to cope with emotional distress
(7). Psychiatric disorders have been conceptualized as chronic distress states associated with neurobiological alterations in brain stress circuits
(8–
10). On the other hand, chronic drug use is associated with neuroadaptations in brain reward pathways that produce secondary psychiatric symptoms during acute and protracted withdrawal states
(10). With increasing severity of addiction, neuroadaptations in stress and reward circuits occur, and these changes may underlie the increasing emotional distress often associated with substance use disorders
(11,
12).
A growing body of evidence from basic science and translational studies implicates common neurobiological pathways and abnormalities involved in addiction and a number of psychiatric disorders. Within a neurobiological framework, at least two hypotheses can be postulated to explain comorbidity: 1) addiction and other psychiatric disorders are different symptomatic expressions of similar preexisting neurobiological abnormalities, and 2) repeated drug administration, through neuroadaptation, leads to biological changes that have common elements with the abnormalities mediating certain psychiatric disorders
(13).
One of the bridging constructs between psychiatric and substance use disorders is the role of stress in the development and relapse of substance use disorders and other psychiatric disorders.
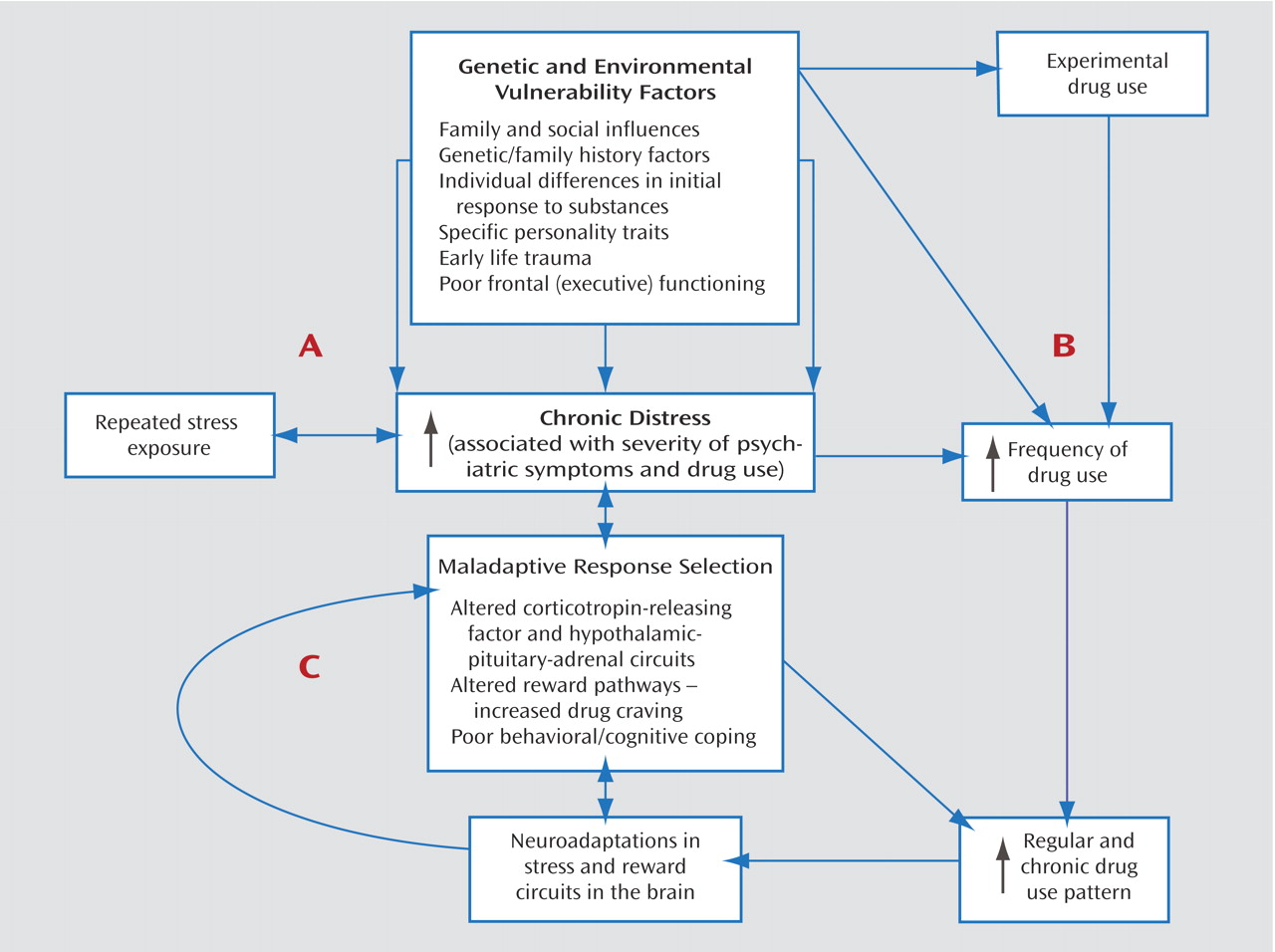
Figure 1 provides a heuristic model of the relationship between chronic distress states, substance use disorders, and psychiatric comorbidity. Although the model conceptualizes chronic distress as the bridging construct, various genetic and environmental vulnerability factors contribute to the development of the distress states, as noted in
Figure 1. Additional research on these factors will contribute to a more specific understanding of the mechanisms underlying the associations between psychiatric and substance use disorders.
Corticotropin-releasing factor (CRF), one of the key hormones involved in the stress response, has been implicated in the pathophysiology of anxiety, affective, and addictive disorders
(8,
14). Preclinical evidence suggests that CRF and noradrenergic pathways are involved in stress-induced reinstatement of drug-seeking behavior in drug-dependent laboratory animals
(15). Stress stimuli that activate CRF circuits are also known to potentiate mesolimbic dopaminergic reward pathways in laboratory animals
(16). Similarly, human laboratory studies have shown that emotional stress and negative affect states increase drug craving in drug-dependent individuals
(17,
18). Evidence of an association between severity of depressive symptoms in patients with major depression and the subjective reinforcing effect of an acute dose of dextroamphetamine
(19) suggests dysregulation of reward systems with increasing levels of distress in major depression. In animal models, early life stress and chronic stress result in long-term changes in stress responses
(20). Such changes can alter the sensitivity of the dopamine system to stress and can increase susceptibility to self-administration of substances of abuse
(16,
21,
22).
Corticolimbic dopamine and noradrenergic pathways modulate prefrontal cortical function under conditions of increasing cognitive or emotional demand, including persistent distress states, tasks involving high levels of cognitive challenge, and working memory tasks
(23,
24). Glutamatergic and γ-aminobutyric acid (GABA)-ergic pathways are also important in modulating prefrontal cortical function
(25).
It is important to note that different substances of abuse have widely varying effects on neurobiological systems. Cocaine and amphetamines have a stimulating effect on catecholaminergic systems. Opioid analgesic drugs act through a complex system of opioid receptors, and nicotine acts through specific nicotinic receptors distributed throughout the central and peripheral nervous systems. GABA-ergic and glutamatergic systems are particularly important in acute intoxication and withdrawal from alcohol and benzodiazepines. Clearly, the effects of acute intoxication and withdrawal differ for each of these drugs, and the effect on psychiatric disorders also differs by drug. It is interesting to note, however, that there appear to be common neurobiological pathways operating across substances of abuse. Dopamine activity in the nucleus accumbens has been implicated in the mechanism of reinforcement for almost all drugs of abuse
(26). Furthermore, drugs of abuse activate the CRF/hypothalamic-pituitary-adrenal (HPA) axis during use/abuse, and alterations in the CRF/HPA and noradrenergic systems during acute withdrawal/abstinence are also well documented
(11,
12,
26). Some animal models of reinstatement (i.e., stress-induced reinstatement, cue-induced reinstatement) operate across substances of abuse, also arguing for some common mechanisms.
In the following sections, we review emerging data that shed light on the neurobiological connections between various substance use disorders and the four psychiatric disorders considered here (depression/mood disorders, PTSD, ADHD, and schizophrenia).
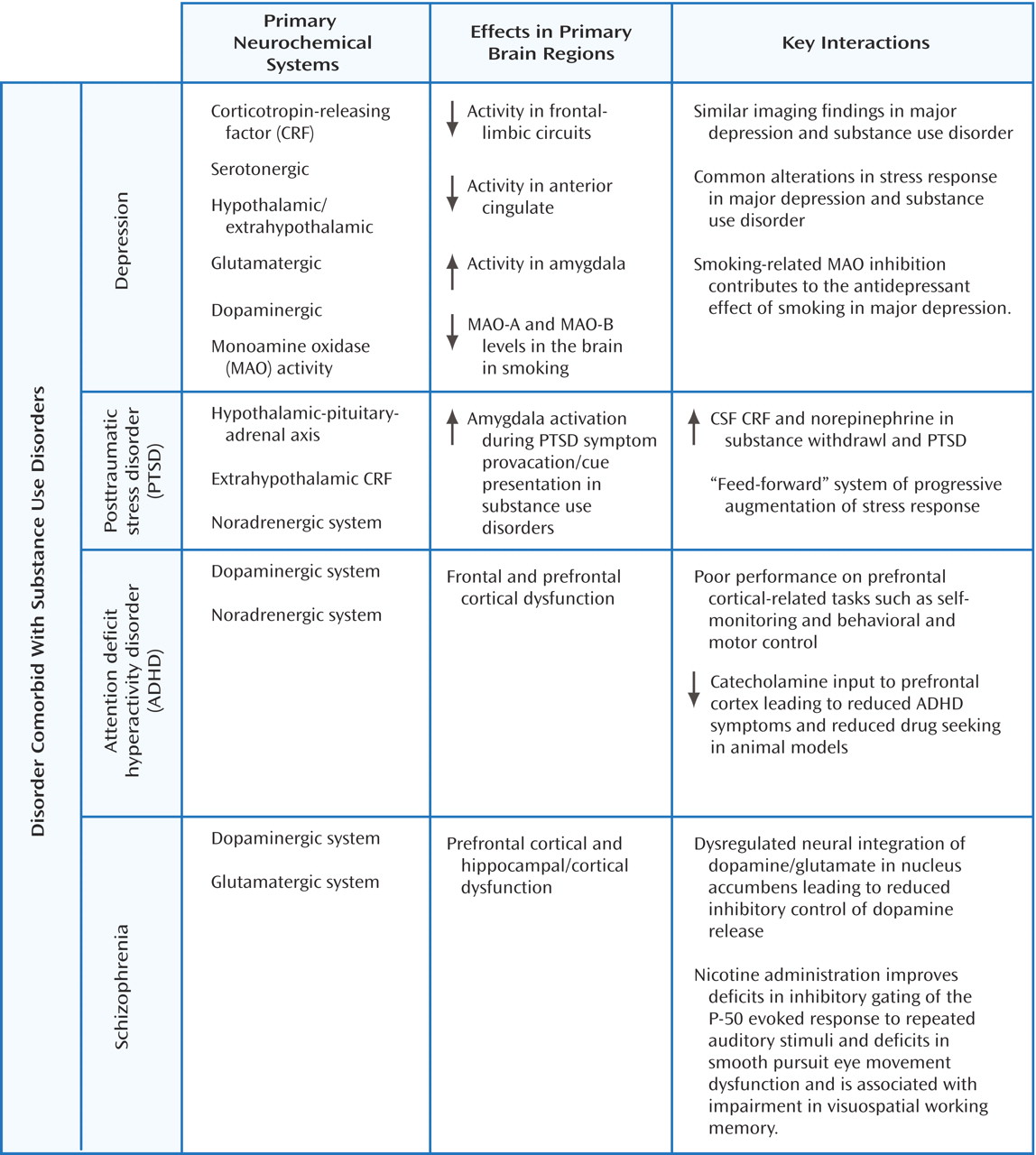
Figure 2 summarizes the neurobiological evidence cited in the following sections and identifies the overlapping neurotransmitter systems and associated brain regions.
Depression and Substance Use Disorders
Epidemiological studies reported rates of comorbidity of major depression with nicotine, alcohol, and illicit drug abuse ranging from 32% to 54%
(1,
27,
28). Individuals with major depression are more likely to develop substance use disorders, and individuals with substance use disorders are at greater risk for the development of major depression, compared to the general population
(27–
29). Clinical similarities exist between major depression and substance use disorders. Depressive symptoms are commonly reported during acute and chronic withdrawal from drugs of abuse. Irritability, sleep difficulties, anxiety, and trouble with attention/concentration are associated with both protracted withdrawal states and major depression.
Neurobiological similarities between major depression and substance use disorders likely contribute to both symptom overlap and high rates of comorbidity
(13). Substantial data indicate that extrahypothalamic CRF and HPA axis abnormalities
(8) and alterations in catecholamine, serotonin, GABA, and glutamate systems are associated with major depression
(30,
31). Neuroadaptations associated with chronic drug abuse are associated with alterations in these neurotransmitter systems, especially during acute withdrawal states
(13). CRF/HPA response during acute drug withdrawal has a positive association with withdrawal-related distress and with depressive symptoms
(32,
33). In addition, a growing amount of evidence indicates that the neurobiological alterations associated with acute withdrawal last for varying time periods and contribute to drug craving and relapse in substance use disorders
(12). In a recent study
(34), individuals with substance use disorders, both with and without depressive symptoms, were found to have significantly lower ACTH and cortisol response to CRF stimulation, compared to healthy subjects. These findings are consistent with studies of abstinent smokers, alcoholic subjects, and subjects with polysubstance dependence in which a blunted cortisol response to standard psychological stressors was demonstrated
(12). Blunted cortisol and prolactin responses to
d-fenfluramine challenge in abstinent heroin-dependent individuals with and without depression have also been reported
(35). Blunted peripheral stress hormone responses may be a marker for increased HPA axis activity
(36,
37).
Evidence of altered neuroendocrine response to stress challenges in substance use disorders is consistent with clinical observations that individuals with substance use disorder have difficulty managing stressful situations and emotional distress states and often relapse in the face of stressful situations
(12,
38). In laboratory studies, stress and negative affect states increase drug craving and emotional distress in abstinent substance-dependent individuals
(17,
39–41). These changes are accompanied by physiological arousal
(18), and this finding suggests that drug-craving states that are marked by increased levels of anxiety and distress are accompanied by biological stress responses. Increased distress-related drug craving is associated with vulnerability to continued drug use and relapse
(12,
42), and this association suggests a mechanistic connection between depressive symptoms and substance use disorders.
In other studies, specific associations between monoamine oxidase (MAO) activity in smoking and major depression have been examined. MAO (with A and B subtypes), an enzyme involved in oxidizing serotonin, norepinephrine, and dopamine in the brain, has long been associated with negative mood and depression. For example, MAO inhibitors are known to have antidepressant properties. It is interesting to note that smokers show reduced MAO-A and MAO-B levels in the brain, compared to nonsmokers and former smokers
(43,
44). These findings provide some support for the notion that smoking may have antidepressant effects through inhibition of MAO-A and MAO-B activity and suggest a pharmacological explanation for the high rates of smoking reported among individuals with major depression.
Recent findings from neuroimaging studies implicate similar alterations in frontal-limbic brain circuitry in substance use disorders and major depression. Reduced frontal metabolism and hypoactivity of the anterior cingulate have been reported in individuals with substance use disorders
(45,
46). Significant reduction in dopamine D
2 receptors, particularly in frontal-striatal regions, has been noted in cocaine- and alcohol-dependent individuals, compared to healthy subjects
(45). Reduced frontal-limbic metabolism has also been found in subjects with major depression, relative to healthy subjects
(47). Such findings are consistent with postmortem studies showing reduced cell density and gray matter volume in individuals with a diagnosis of major depression
(48). Furthermore, amygdala hyperactivity and anterior cingulate hypoactivity are associated with major depression
(47), and studies of individuals with substance use disorders indicate activation in the amygdala associated with cue-induced drug craving
(49,
50). Under conditions of distress, cocaine-dependent subjects exhibited decreased activity in frontal regions such as the medial prefrontal cortex and the anterior cingulate, similar to that seen with negative mood in subjects with major depression
(51,
52). Similarly, a recent study
(53) reported lower levels of glucose metabolism in the anterior cingulate and insula, but higher levels in the orbitofrontal region, amygdala, middle and posterior cingulate, and ventral striatum in methamphetamine abusers with severe mood and anxiety symptoms, compared to healthy subjects.
In conclusion, neuroendocrine and neuroimaging studies indicate dysregulation in frontal-limbic systems associated with stress and reward pathways in both major depression and substance use disorders. This common dysregulation is likely to contribute to the high rate of comorbidity of these illnesses. Evidence concerning negative affect and stress-related drug seeking/craving provides additional insight into emotional distress states and drug use in drug-experienced individuals. A better understanding of these connections will contribute to the development of new treatments for major depression, substance use disorders, and the comorbidity of these disorders.
PTSD and Substance Use Disorders
The high prevalence of the comorbidity of substance use disorders and PTSD has been reported in a number of studies. Initial reports focused on veterans with PTSD, of whom 64%–84% met the criteria for a lifetime alcohol use disorder and 40%–44% met the criteria for a lifetime drug use disorder, including nicotine dependence
(54,
55). In civilian populations with PTSD, estimates of the lifetime prevalence of substance use disorders range from 22% to 43%
(56,
57), far higher than the estimates for substance use disorders in the general population.
As in other comorbidities, PTSD and substance use disorders have a number of connecting pathways. Substance intoxication may heighten the likelihood of exposure to trauma, hence the likelihood of developing PTSD. Furthermore, chronic substance use and withdrawal may increase anxiety/arousal states, making it more likely that individuals with substance use disorders will develop PTSD after trauma exposure. On the other hand, PTSD could increase the risk of developing a substance use disorder, because individuals may abuse substances in an attempt to relieve symptoms of PTSD. Substance use could also exacerbate symptoms and/or prolong the course of PTSD by preventing habituation to traumatic memories. These pathways are not mutually exclusive, and new evidence is emerging concerning the neurobiological underpinnings of potential causal pathways. In one recent study
(58), individuals who had experienced any trauma and developed PTSD had an increased risk for the development of drug dependence, particularly nicotine dependence, but not alcohol dependence. This finding suggests specificity between substance of abuse and psychopathology.
The HPA axis, extrahypothalamic CRF, and the noradrenergic system are all intimately involved in the stress response, PTSD, and the pathophysiology of substance use disorders. Evidence is accumulating to support a role for CRF in mediating the effects of stress in increasing self-administration of drugs. Studies in rats have also demonstrated that withdrawal from chronic cocaine
(59) or alcohol administration
(60) in rats is associated with increases in CRF in the hypothalamus, amygdala, and basal forebrain. Elevated CSF CRF has been found in humans during alcohol withdrawal
(61). Two studies examining CSF concentrations of CRF have demonstrated higher levels in individuals with PTSD, compared to healthy subjects
(62,
63). This finding is of particular interest because elevated brain CRF levels, especially in the amygdala, potentiate fear-related behavioral responses
(64). As such, elevated levels of CRF may mediate both the symptoms of hyperarousal and the increased risk for substance use disorders in PTSD. Increased CRF may enhance the reinforcing properties of some drugs, worsen the severity of withdrawal symptoms, and exacerbate symptoms of PTSD.
Evidence implicating abnormalities in noradrenergic systems has been found for both PTSD and substance use disorders. Individuals with PTSD have elevated urinary excretion of both norepinephrine and epinephrine and elevated plasma levels of norepinephrine
(65). Markers of noradrenergic activity are increased in both alcohol and opioid withdrawal
(66–
68). Brain CRF and noradrenergic systems modulate each other in a number of ways. Stress increases CRF in the locus ceruleus
(69), and intraventricular administration of CRF increases norepinephrine turnover in the hypothalamus, hippocampus, and prefrontal cortex
(70). In the amygdala, norepinephrine stimulates the release of CRF
(71). Koob and colleagues
(72,
73) hypothesized that interactions between CRF and the noradrenergic systems can function as a “feed-forward” system, with progressive augmentation of the stress response with repeated stress exposure. Specifically, substance use or withdrawal or other stress may stimulate CRF release in the locus ceruleus, leading to the release of norepinephrine in the cortex, which would, in turn, stimulate the release of CRF in the hypothalamus and amygdala. This interaction could help to explain the attempt to self-medicate PTSD symptoms with substances of abuse, the worsening of PTSD symptoms during substance withdrawal, and the increase in vulnerability to the development of PTSD in traumatized individuals with substance use disorders.
Neuroimaging studies have shed light on the connection between PTSD, other anxiety disorders, and substance use disorders. Amygdala activation occurs during symptom provocation in PTSD, panic disorder, and social phobia
(74). As mentioned earlier, increased amygdalar blood flow is also seen in cocaine-dependent individuals presented with cocaine-related cues
(50,
75).
ADHD Spectrum and Substance Use Disorders
Substantial evidence suggests that ADHD, conduct disorder, and oppositional defiant disorder co-occur at high rates among children and adolescents. This group of disorders, conceptualized as externalizing disorders, is associated with shared genetic and environmental risk factors
(76–
78). Externalizing disorders are commonly comorbid with substance use disorders in adolescents, with prevalence estimates ranging from 30%–50%
(79). Adolescents with comorbid substance use disorder and ADHD, conduct disorder, and/or oppositional defiant disorder have an earlier age at onset and a more severe course of substance use disorder
(80–
82). Research has identified genetic, neurobiological, and psychosocial risk factors that contribute to the core pathophysiology in the development of comorbid ADHD and substance use disorders.
Externalizing disorders are characterized by behavioral disinhibition and personality traits such as aggression, high levels of impulsivity, and poor self-control
(77,
78,
83). Substantial evidence suggests that externalizing disorders are associated with problems in higher-order “executive” (frontal) cognitive function. In fact, ADHD has often been characterized as a disorder of frontal and prefrontal cortex dysfunction
(23,
71). Children with ADHD, conduct disorder, oppositional defiant disorder, and early-onset substance use disorder showed poor performance on neuropsychological tests of abilities involving the prefrontal cortex, including planning, attention, cognitive flexibility, working memory, self-monitoring, and behavioral and motor control
(71).
In a large twin study that examined P3 amplitude—a robust electrophysiological marker with a strong genetic basis—lower P3 amplitude was associated with presence of ADHD, conduct disorder, oppositional defiant disorder, and substance use disorder in adolescent boys
(78). Lower P3 amplitude at age 17 years predicted development of substance use disorder at age 20 years. Although genetic factors contribute to the development of comorbid ADHD, conduct disorder, and oppositional defiant disorder, a single shared environmental factor, identified as parent-child conflict (i.e., negative social interactions between parents and children), accounts for an even larger proportion of the variance
(84). Negative parent-child interactions, high levels of negative affect, and emotional distress are also known to increase the risk of substance use disorder in adolescents
(85). These data suggest that coping with high levels of family conflict may play an important role in the development of both ADHD and substance use disorders.
Preclinical research has demonstrated that dopamine and norepinephrine modulate prefrontal cortical function
(23,
86) and that stress impairs prefrontal cortical function
(86,
87). Evidence from brain imaging studies indicates that the prefrontal cortex and anterior cingulate cortex play important roles in cognitive conflict monitoring
(88,
89) and self-regulation processing
(90,
91). Difficulties in response inhibition and self-regulation are core symptoms of externalizing disorders
(78,
83). Compared to healthy subjects, boys with comorbid ADHD, conduct disorder, and oppositional defiant disorder show greater behavioral aggression, heart rate reactivity, and higher levels of anger in a laboratory-induced provocation paradigm
(92). Furthermore, in boys with externalizing symptoms, lower cortisol levels and the personality traits of low levels of self-control and harm avoidance are associated with the development of substance use disorders
(93,
94). Thus, consistent with preclinical evidence indicating that stress impairs prefrontal cortical function, human studies suggest that individuals with ADHD and early-onset substance use disorders have poor stress-related coping and poor self-regulation.
The prefrontal cortex and anterior cingulate cortex are also important in regulating behavior related to future rewards. Primate studies have shown that the prefrontal cortex and anterior cingulate cortex are involved in assessing reward expectancy
(95) and motor responses based on future reward
(96). These data are consistent with the critical role of the prefrontal cortex in drug self-administration and in the reinforcement and reinstatement of drug use
(97,
98). Children with ADHD and conduct disorder show disinhibited physiological and behavioral responses during reward-related cognitive tasks
(99,
100). These findings are consistent with decreased prefrontal cortical and striatal activity and increased activity in posterior and sensory cortices in ADHD
(101,
102) that is normalized by chronic methylphenidate treatment
(103). Preliminary findings indicated that decreasing catecholamine input to the prefrontal cortex by means of α
2-adrenergic agonists such as guanfacine, which inhibit norepinephrine centrally, enhances prefrontal cortical function and decreases ADHD symptoms
(104). It is interesting to note that other α
2-adrenergic agonists, such as clonidine and lofexidine, attenuate stress-induced reinstatement of drug-seeking behavior in laboratory models
(105,
106). To the extent that stress and reward dysfunction contribute to prefrontal cortical deficits in ADHD and substance use disorders, α
2-adrenergic agonists may be beneficial in addressing this comorbidity.
Schizophrenia and Substance Use Disorders
Recent studies have demonstrated that up to 50% of individuals with schizophrenia have either alcohol or illicit drug dependence and more than 70% are nicotine dependent
(2,
107,
108). In addition to having the expected adverse medical consequences, substance use in schizophrenic patients is associated with poor social function, symptom exacerbation, frequent hospitalization, medication noncompliance, and poor treatment response
(109,
110). Schizophrenia and substance use are connected by multiple potential links, including genetic vulnerability, medication side effects, negative symptoms, and psychosocial factors. Self-medication has been commonly invoked to explain the high comorbidity. Specifically, self-medication of negative symptoms, such as social withdrawal and apathy, and drug use in the attempt to decrease discomfort from the side effects of typical antipsychotic medications have been suggested as explanations for the high prevalence of substance use disorders in individuals with schizophrenia. Although these factors may play some role, advances in neurobiology suggest that the neuropathology of schizophrenia affects the neural circuitry mediating drug reward, leading to an increased vulnerability to addiction. Specifically, Chambers and colleagues
(111) hypothesized that abnormalities in hippocampal-cortical function in schizophrenia impair the inhibitory hippocampal projections to the nucleus accumbens, resulting in reduced inhibitory control over dopamine-mediated functional hyperresponsivity to dopamine release. In this model, dysregulated neural integration of dopamine and glutamate in the nucleus accumbens resulting from frontal and hippocampal dysfunction could lead, in subjects without prior drug exposure, to neural and motivational changes similar to those in long-term substance use. Thus, the predilection of schizophrenic patients to substance use disorders may be a primary disease symptom.
Recent studies focused on the neurobiological interface between schizophrenia substance use disorders support this hypothesis. In one study
(112), magnetic resonance images in groups of subjects with schizophrenia, schizophrenia plus alcohol dependence, and alcohol dependence only were compared with those from a matched control group. Gray matter deficits were found in all three patient groups, but were greatest in the group with comorbidity. The most prominent deficits were in the prefrontal and anterior superior temporal regions, indicating that comorbidity compounded the prominent prefrontal cortical deficits that are present independently in schizophrenia and alcohol dependence. Lifetime alcohol consumption in subjects with comorbidity was approximately five times less than that in the alcohol-dependent subjects, yet the subjects with comorbidity exhibited the full detrimental effects of alcohol, which suggests an interactive effect.
One area of particular interest is nicotine dependence and schizophrenia. It has been estimated that 70%–90% of individuals with chronic schizophrenia are nicotine dependent
(113). Nicotine interacts with many of the same central pathways involved in schizophrenia, including the dopaminergic and glutamatergic pathways in the mesolimbic areas. Several abnormalities associated with schizophrenia are improved with nicotine administration, including deficits in the inhibitory gating of the P-50 evoked response to repeated auditory stimuli
(114) and deficits in smooth pursuit eye movement dysfunction
(115). George and colleagues
(116) found deficits in visuospatial working memory in schizophrenic and nonschizophrenic individuals with nicotine dependence. With increasing periods of abstinence, the nonschizophrenic smokers had improvements in visuospatial working memory, whereas the schizophrenic smokers experienced further impairment in visuospatial working memory. The authors postulated that the high rates of cigarette smoking in schizophrenic patients may be related to the effects of smoking in alleviating some of the cognitive dysfunction associated with the presumed hypofunctionality of cortical dopamine systems in schizophrenia. In a recent study of more than 14,000 adolescents followed over a 4–16-year period, adolescents who smoked more than 10 cigarettes/day at the initial evaluation were significantly more likely to be hospitalized for schizophrenia during the follow-up period
(117). These findings suggest that smoking might constitute self-medication of premorbid symptoms, might reflect an intrinsic, disease-related disorder of nicotinic transmission, or might play a causative role in the development of schizophrenia through chronic activation of mesolimbic dopaminergic neurotransmission in vulnerable individuals. The development of novel approaches based on nicotinic receptor mechanisms may have implications for both prevention and treatment of schizophrenia.
Data from small, largely uncontrolled studies suggest that treatment with clozapine and other atypical antipsychotics may be associated with decreases in substance abuse in schizophrenic patients. Although the data are limited, this favorable response to atypical agents is consistent with the theory that dysfunction of the brain reward system leads to an increased vulnerability to addictions in schizophrenia
(118). Typical antipsychotic agents are potent antagonists of D
2 receptors. Although this blockade may initially decrease the reinforcing properties of some substances of abuse, with chronic use there may be enhancement of the substances’ reinforcing properties. Studies in rodents demonstrated that chronic treatment with haloperidol increased the reinforcing properties of cocaine, presumably through up-regulation of the postsynaptic dopamine receptor secondary to chronic blockade
(119). In contrast, atypical agents, such as clozapine, have varied actions on a number of neurotransmitter systems and are much weaker D
2 antagonists. It is possible that these agents have a normalizing effect on the signal detection capabilities of the mesocorticolimbic reward circuitry, and this action may explain the association with decreased substance use.
Conclusions
Although the nature of the relationship between psychiatric disorders and substance use disorders is complex and multifaceted, there are likely to be unifying constructs. Neuroadaptations in brain stress and reward pathways associated with chronic stress may predispose or unmask a vulnerability to psychiatric disorders, substance use disorders, or both. Dysfunction in the prefrontal cortex and frontal cortex associated with deficits in self-monitoring and behavioral control are evident in ADHD, other externalizing disorders, and substance use disorders. Emerging evidence suggests that abnormalities of glutamatergic function in schizophrenia and other psychiatric disorders may mediate vulnerability to the development of substance use disorders.
Although the focus of this article has been on neurobiological connections between psychiatric and substance use disorders, it is important to note that these connections constitute just one facet of a complex issue. Further exploration of overlapping neural circuitry and mechanistic relationships will be essential in guiding treatment and prevention efforts. However, improvement in our understanding of co-occurring disorders will be useful only if there is a treatment system in place to implement these findings. Clearly, change at public policy levels will be necessary to maximize the benefits derived from the findings of neurobiological explorations in order to improve the lives of individuals with comorbidity.