Atomoxetine is a highly selective inhibitor of the noradrenergic transporter and was approved in the United States in November 2002 for the treatment of attention deficit hyperactivity disorder (ADHD). Results from premarketing trials demonstrated effect sizes between 0.6 and 0.8
(1 –
4) . However, these trials did not include comparisons to stimulants suitable for assessing differential response and tolerability. Such comparisons are important because stimulants, such as methylphenidate, are well established and highly effective interventions for ADHD
(5) . In addition, it is important to evaluate the extent to which there may be differential response to the two classes of medications, since not all individuals tolerate or respond optimally to stimulants.
Four published reports have compared responses to stimulants and atomoxetine in the same study, but all have important limitations. We found that responses to atomoxetine and methylphenidate were not different
(6), but interpretation of those results is constrained by the facts that the study was not placebo controlled, the methylphenidate group was small (atomoxetine, N=184; methylphenidate, N=44), and methylphenidate was administered as an immediate-release formulation given one to three times per day. Sangal et al.
(7) examined sleep latency (and secondarily, clinical response) in a crossover study of atomoxetine and immediate-release methylphenidate in 85 children (75 completers) with ADHD. Responses to the two treatments were again comparable. However, this study was primarily intended to assess sleep, not clinical response, so it is possible that certain demographic or clinical characteristics of the participants were different from what would be expected in more traditional ADHD efficacy trials.
Two other comparator studies used long-acting stimulant formulations. This is relevant because the majority of children in the United States treated with stimulants receive new, longer-acting formulations. Kemner et al.
(8) conducted a large community-based, open-label study in children 6 to 12 years of age with ADHD (N=1,323), who were treated for 3 weeks with either once-daily osmotic-release oral methylphenidate, a long-acting methylphenidate formulation (N=850), or atomoxetine (N=473). Treatment with either medication significantly reduced ADHD symptoms, as measured by the ADHD Rating Scale
(9) ; the mean change scores were 20.24 for osmotically released methylphenidate and 16 for atomoxetine, a statistically significant difference favoring methylphenidate. However, interpretation of the findings is constrained by the open design, the manner in which subjects were assigned to treatments, the unequal cell sizes, the short duration of treatment (possibly inadequate for accurate assessment of response to atomoxetine), and relatively low dosing of atomoxetine. Finally, Wigal et al.
(10) compared response to extended-release mixed amphetamine salts and atomoxetine in 203 subjects in a 3-week laboratory classroom design. Results on several measures favored extended-release mixed amphetamine salts. However, the short duration of the study and substantially different approach to evaluating treatment response (i.e., different assessment measures, laboratory classroom design) makes it difficult to contrast these findings with those of the other comparator studies. Moreover, response to atomoxetine in this study was relatively poor and considerably different from that in other studies, raising questions regarding generalizability of the findings.
There are virtually no data regarding the extent to which there is differential response to stimulants and atomoxetine. In the Sangal et al. study
(7) of immediate-release methylphenidate and atomoxetine, 36% of the subjects (χ
2 =0.06) responded preferentially to one or the other treatment. This trend, suggesting preferential response in a subset of subjects, is consistent with the practice guideline of the American Academy of Child and Adolescent Psychiatry
(11), which advocates using a second medication class if treatment with the first is not successful. However, results from this study are not sufficient to establish this as an evidenced-based practice; thus, determining the number of preferential responders to atomoxetine and stimulants in a larger sample would be of considerable clinical value. In particular, it would be useful to determine how many subjects who do not respond to stimulant treatment can be successfully treated with atomoxetine because stimulants are still often the first treatment used and many patients do not achieve optimal response or tolerability
(12,
13) .
To compare the acute treatment response of children and adolescents with ADHD to atomoxetine and methylphenidate, we designed a large parallel-group, placebo-controlled, randomized study using osmotically released methylphenidate, the most frequently used and longest-acting methylphenidate formulation (IMS Health, IMS National Prescription Audit Plus, February 2006), as the comparator. Following the acute comparison trial, all of the subjects initially treated with osmotically released methylphenidate who remained in the study were crossed over to receive atomoxetine under double-blind conditions. This provided an opportunity to collect preliminary data regarding differential response to the two treatments.
Method
Participants
The patients were children and adolescents, ages 6 to 16 years, who met the DSM-IV criteria for ADHD, any subtype, as determined by clinical history and confirmed by a semistructured interview, the Schedule for Affective Disorders and Schizophrenia for School Aged Children—Present and Lifetime Version (K-SADS-PL)
(14) . Symptom severity at entry was required to be at least 1.5 standard deviations above the U.S. age and gender norms as assessed by the ADHD Rating Scale-IV—Parent Version: Investigator-Administered and -Scored
(9) . Assessment of concurrent psychiatric diagnoses was made by clinical interview and confirmed by the K-SADS-PL. Patients who had seizures, bipolar disorder, a psychotic illness, or a pervasive developmental disorder or who were taking concomitant psychoactive medications were excluded from the study. Because anxiety and tic disorders are relative contraindications for use of osmotically released methylphenidate (according to the product label), patients with these conditions were also excluded. Other concurrent psychiatric diagnoses, including major depressive disorder, were permitted as long as ADHD was the primary diagnosis and therefore an appropriate target of treatment.
Subjects could either have been treated previously with stimulants or be treatment naive. However, for ethical reasons subjects were excluded if they had been treated previously with an adequate trial of methylphenidate or amphetamine and either did not experience at least some improvement in ADHD signs and symptoms (nonresponders) or had intolerable adverse events. An adequate trial was defined as lasting at least 4 weeks and reaching a total daily dose of at least 1.0 mg/kg per day or 60 mg/day (whichever was lower) of immediate-release methylphenidate, 36 mg/day of osmotically released methylphenidate, or 0.5 mg/kg per day of d -amphetamine or mixed amphetamine salts.
The study was conducted at 20 sites in the United States. After oral description of the study, written informed consent was obtained from a parent or guardian for each patient, and each youth provided written assent. The study was reviewed and approved by each site’s ethical review board and was conducted in accordance with the ethical standards of the 1975 Declaration of Helsinki as revised in 2000
(15) and all applicable regulatory requirements.
Measures
The primary efficacy (response) measure was the investigator-administered and -scored version of the ADHD Rating Scale, a validated 18-item scale with well-described psychometric properties
(9) . Ratings on this instrument, scored by the clinician following a semistructured interview with the parent, were used to measure ADHD symptom severity in the past week and were obtained for items corresponding to each of the 18 behavioral descriptors of ADHD in DSM-IV. Item scores ranged from 0 (never or rarely) to 3 (very often). “Response” was defined as a decrease from baseline of 40% or more in the total ADHD Rating Scale score at week 6. This threshold was specified a priori in the statistical analysis plan and protocol. It was chosen on the basis of the degree of change in the ADHD Rating Scale score that most often corresponded to a clinically meaningful atomoxetine response in previous trials, as defined by a rating of 1 or 2 (i.e., no symptoms or minimally symptomatic) for ADHD on the Clinical Global Impression (CGI) severity scale. The relevance of this criterion is further supported by an analysis by Gao et al.
(16), and it falls in the range of categorical criteria used to define response in studies of methylphenidate using the ADHD Rating Scale that have been conducted and reported by other investigators
(17,
18) .
The secondary outcome measures included the CGI ADHD severity scale, Conners Parent Rating Scale ADHD index
(19), Daily Parent Ratings of Evening and Morning Behavior—Revised (a short rating scale that was completed by the primary caregiver to rate the child’s out-of-school behaviors each day during a 5-day period), and the Child Health Questionnaire, a validated measure of functional outcomes and quality of life
(20) . At the outset of the trial, all raters were trained in the use of the study instruments by observing live and video interviews.
Each participant had a complete medical evaluation at baseline that included a physical examination and the following laboratory measures: routine blood chemistry, liver function tests, hematologic measures, urinalysis, urine drug screen, and ECG. All of these laboratory measures except the drug screen were repeated during each study period. Safety was assessed at each visit by open-ended questioning for adverse events and by measurement of vital signs.
Study Design
The acute comparison trial was a 6-week randomized, double-blind, placebo-controlled parallel-design study comparing atomoxetine and osmotically released methylphenidate. Patients were required to discontinue any psychoactive medication for at least five times the medication’s plasma half-life (or at least 5 days) before entering the study. After two pretreatment assessment visits, patients were randomly assigned to receive one of three treatments: atomoxetine (0.8–1.8 mg/kg per day, administered as a divided twice-daily dose), osmotically released methylphenidate (18–54 mg/day, administered as a single morning dose), or placebo. The randomization ratio was 3:3:1 for atomoxetine, osmotically released methylphenidate, and placebo, respectively. Treatment was initiated at a standard specified dose for all patients and could be increased up to the maximum allowable dose by following a predetermined titration schedule, based on the investigator’s judgment of clinical response and tolerability. Postrandomization visits were conducted at weeks 1, 3, and 6. Atomoxetine was initiated at a dose of 0.8 mg/kg, with increases to 1.2 mg/kg per day allowed at day 5 and 1.8 mg/kg per day at the third postrandomization visit. Osmotically released methylphenidate was initiated at 18 mg/day, with increases to 36 mg and 54 mg allowed at the first and second postrandomization visits, respectively. Study doses were chosen to reflect either the product label (in the case of osmotically released methylphenidate) or the doses used in most clinical trials (in the case of atomoxetine, which did not have a label at the time). After this study, osmotically released methylphenidate was approved for use in doses up to 72 mg/day in adolescents, and the maximum atomoxetine labeled dose was set at 1.4 mg/kg per day
(21) .
The study drugs were administered by using a double-dummy design. Patients in each treatment arm took three identically appearing capsules consisting of atomoxetine, osmotically released methylphenidate, or placebo in the morning and two capsules consisting of atomoxetine or placebo in the evening (total of five capsules). The dosing of osmotically released methylphenidate used 18-mg capsules only, each of which was overencapsulated for blinding purposes. Dissolution studies were conducted before the beginning of the trial to ensure that drug release was not adversely affected.
The mean final administered dose for atomoxetine was 1.45 (SD=0.32) mg/kg per day or 53.0 (SD=17.0) mg/day (range: 0.65–1.86 mg/kg per day), which is slightly above the subsequently established labeled dose (1.4 mg/kg per day). The mean final administered dose for osmotically released methylphenidate was 39.9 (SD=14.6) mg/day or 1.16 (SD=0.55) mg/kg per day (range: 0.32–2.57 mg/kg per day). In patients younger than 12 years (N=162), the mean final methylphenidate dose was 39.2 (SD=14.9) mg/day or 1.26 (SD=0.58) mg/kg per day (range: 0.32–2.57 mg/kg per day). In adolescents age 12 or older (N=57) the mean final methylphenidate dose was 41.7 (SD=13.7) mg/day or 0.88 (SD=0.35) mg/kg per day (range: 0.32–1.90 mg/kg per day).
After completion of the initial 6-week comparison trial, subjects originally assigned to receive osmotically released methylphenidate were switched under double-blind conditions to atomoxetine for 6 weeks. This provided exploratory data regarding the extent to which there may be differential response to the two treatments. There was no washout period between the acute and crossover treatment phases. Atomoxetine was administered in the crossover period according to the same dosage titration schedule as in the acute phase of the study. Subjects who were randomly assigned to atomoxetine did not switch to methylphenidate after the acute phase; instead, they entered other studies to evaluate the impact of dose adjustment on response. Double-blind conditions were therefore maintained, because treatments were manipulated after the acute, double-blind comparison phase for all subjects, not just those who were originally assigned to methylphenidate. In addition, both the site investigators and subjects were blinded to the response criterion used in the initial trial and to when that phase ended and the next phase began. These design features all served to protect the blind during the crossover phase of the study.
Statistical Methods
The protocol-specified primary objective was the comparison of response rates in patients treated with atomoxetine and those receiving osmotically released methylphenidate, as measured by the ADHD Rating Scale total score. Primary analyses were performed by using the intent-to-treat group. Response rates were analyzed by using Fisher’s exact test. The number needed to treat (NNT) was also calculated for each treatment in relation to placebo and for atomoxetine in relation to methylphenidate. The number of subjects was chosen to have 90% power to declare noninferiority on the basis of a comparison of response rates, with a noninferiority margin of 15%. The margin of 15% was based on the estimated difference between methylphenidate and placebo seen in two previous atomoxetine studies
(4,
22) using criteria set by regulatory authorities for equivalence studies
(23) . Additional prospectively planned analyses specified in the protocol included response rates in subgroups of previously stimulant-treated and stimulant-naive patients, as well as in extensive and poor cytochrome P2D6 (CYP2D6) metabolizers.
A logistic model with effects for prior stimulant use, investigative site, interaction between treatment and prior stimulant use, and interaction between treatment and investigative site was additionally used to examine response rates. For continuous efficacy measures, change from baseline to endpoint was computed for all patients with baseline and at least one postbaseline measurement, by using a last-observation-carried-forward approach. These variables were analyzed by means of analysis of covariance (ANCOVA) with independent effects for treatment, investigative site, CYP2D6 metabolism status, and baseline score. For binary measures, such as the percentage of patients with treatment-emergent adverse events, pairwise treatment comparisons were conducted by using Fisher’s exact test. Unadjusted p values are reported. Effect size (Cohen’s d) was computed by first calculating the change score for each treatment (change from baseline to endpoint in ADHD Rating Scale total score) and then dividing the difference in change scores between active drug and placebo by the pooled standard deviation (root mean square error). Analyses of safety measures were restricted to randomly assigned patients who took at least one dose of the study drug. All analyses for this article were generated by using SAS/STAT software, version 8 of the SAS system for Windows (SAS Institute, Cary, N.C.).
Results
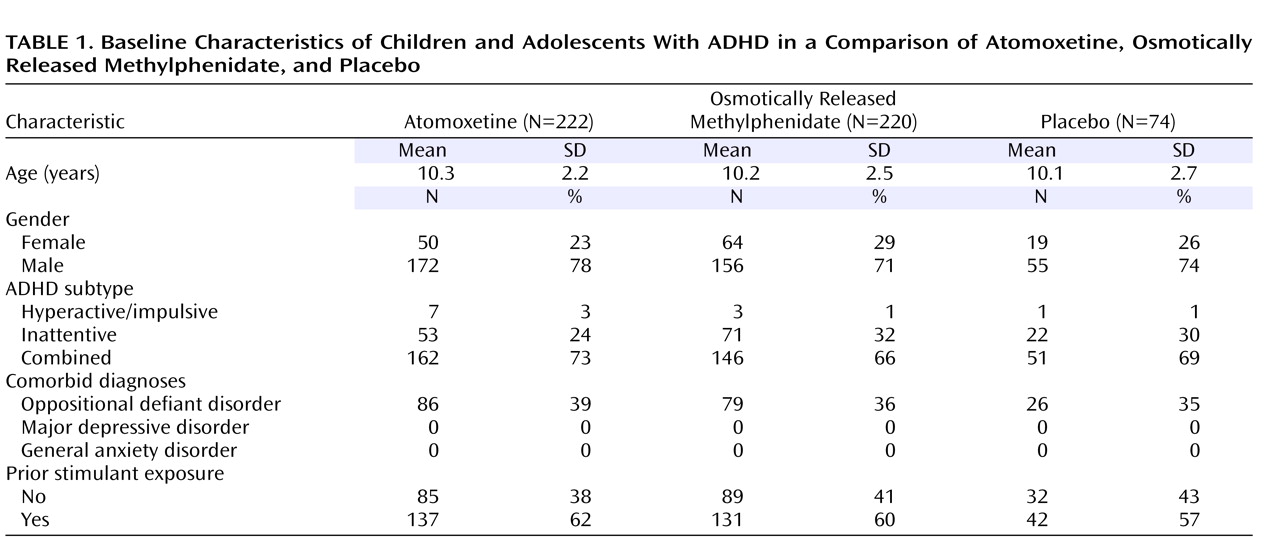
Of 635 patients initially assessed, 516 met the criteria for study entry and were randomly assigned to receive protocol treatment. Patient characteristics (
Table 1 ) and baseline symptom measures (
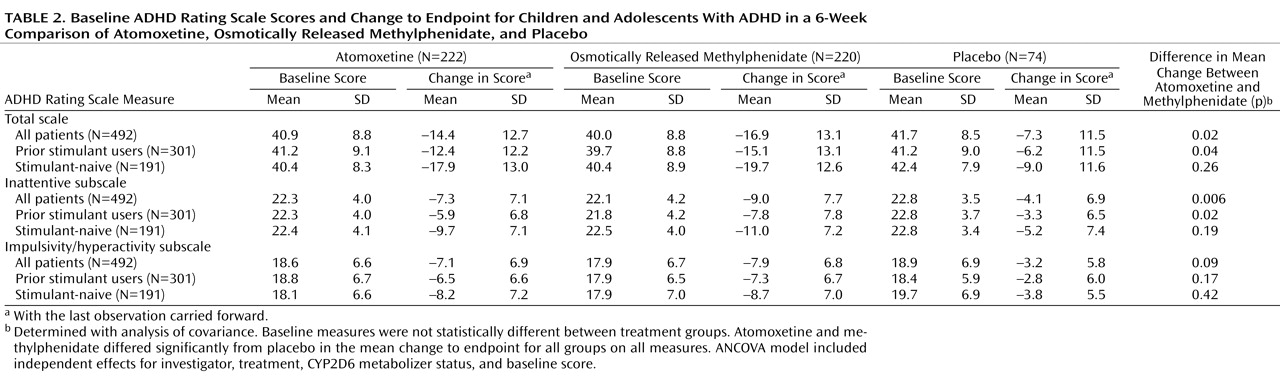
Table 2 ) were similar for all treatment groups.
Assessment 1: Acute Comparison Trial
After 6 weeks of treatment, the proportion of patients responding to atomoxetine (45%) was significantly higher than the rate for placebo (24%) (p=0.003, Fisher’s exact test; NNT=5), and osmotically released methylphenidate (56%) was also superior to placebo (24%) (p≤0.001; NNT=3). In addition, osmotically released methylphenidate was superior to atomoxetine (p=0.02; NNT=9). The 95% confidence interval (CI) for the difference in response rates between atomoxetine and methylphenidate (–21% to –2%) did not support the hypothesized noninferiority of atomoxetine within the protocol-specified 15% threshold. A post hoc analysis found a statistically significant (p<0.05) effect for both drugs in relation to placebo at week 3.
For patients previously treated with a stimulant (N=301), the response rate for osmotically released methylphenidate (51%, p=0.002, Fisher’s exact test; NNT=4), but not atomoxetine (37%, p=0.09; NNT=7), was superior to the rate for placebo (23%), and the response rate for methylphenidate was superior to that for atomoxetine (p=0.03; NNT=8). In patients who were stimulant naive at study entry (N=191), the response rates for both atomoxetine (57%, p=0.004; NNT=4) and methylphenidate (64%, p≤0.001; NNT=3) were superior to the rate for placebo (25%), and the response rates for methylphenidate and atomoxetine were not significantly different (p=0.43, NNT=14). There was not a statistically significant interaction between treatment and previous stimulant use; however, this may be due to the smaller number of patients in this subanalysis (resulting in lower power to detect differences). A similar result was seen in the logistic regression modeling of response rate, where no interaction terms were statistically significant. There were no significant effects associated with investigative site.
Similar results were observed for change in the ADHD Rating Scale total score, as well as scores on the inattentive and hyperactive/impulsive subscales (
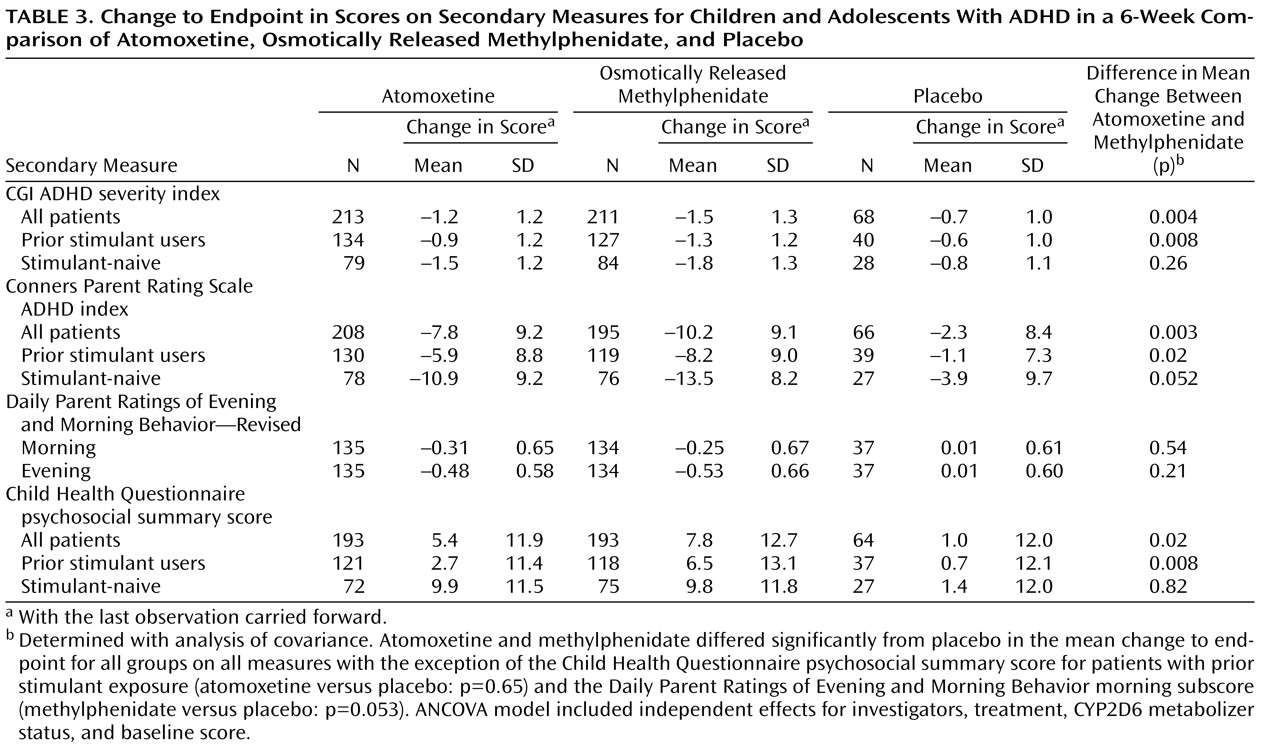
Table 2 ). Treatment effect sizes for the change in total score, in relation to placebo, were 0.6 for atomoxetine and 0.8 for osmotically released methylphenidate. In patients previously treated with stimulants, the treatment effect sizes were 0.5 for atomoxetine and 0.8 for methylphenidate, while in stimulant-naive patients the effect sizes were 0.9 for atomoxetine and 1.0 for methylphenidate. Results of the analyses using secondary measures were similar to those observed for the primary outcome measure (
Table 3 ).
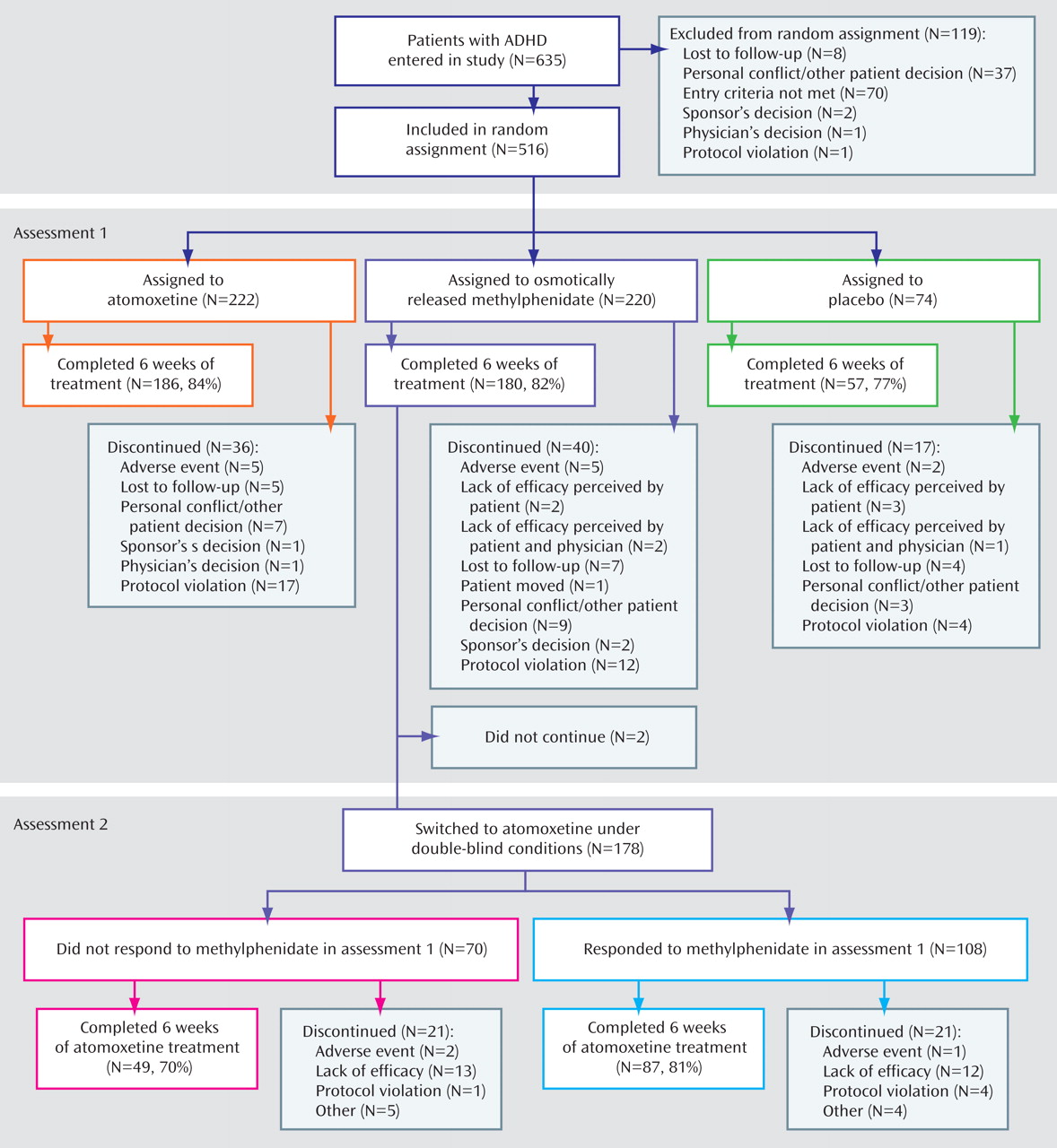
We did not find significant group differences in completion rates for the acute phase of the trial (atomoxetine: 186 of 222, or 84%; osmotically released methylphenidate: 180 of 220, or 82%; placebo: 57 of 74, or 77%; p=0.42, Fisher’s exact test). In addition, the rates of discontinuation due to adverse events were low and similar for all treatments (atomoxetine: five of 222, or 2%; osmotically released methylphenidate: five of 220, or 2%; placebo: two of 74, or 3%; p=1.00). For a more complete description of patient flow through the study, see
Figure 1 .
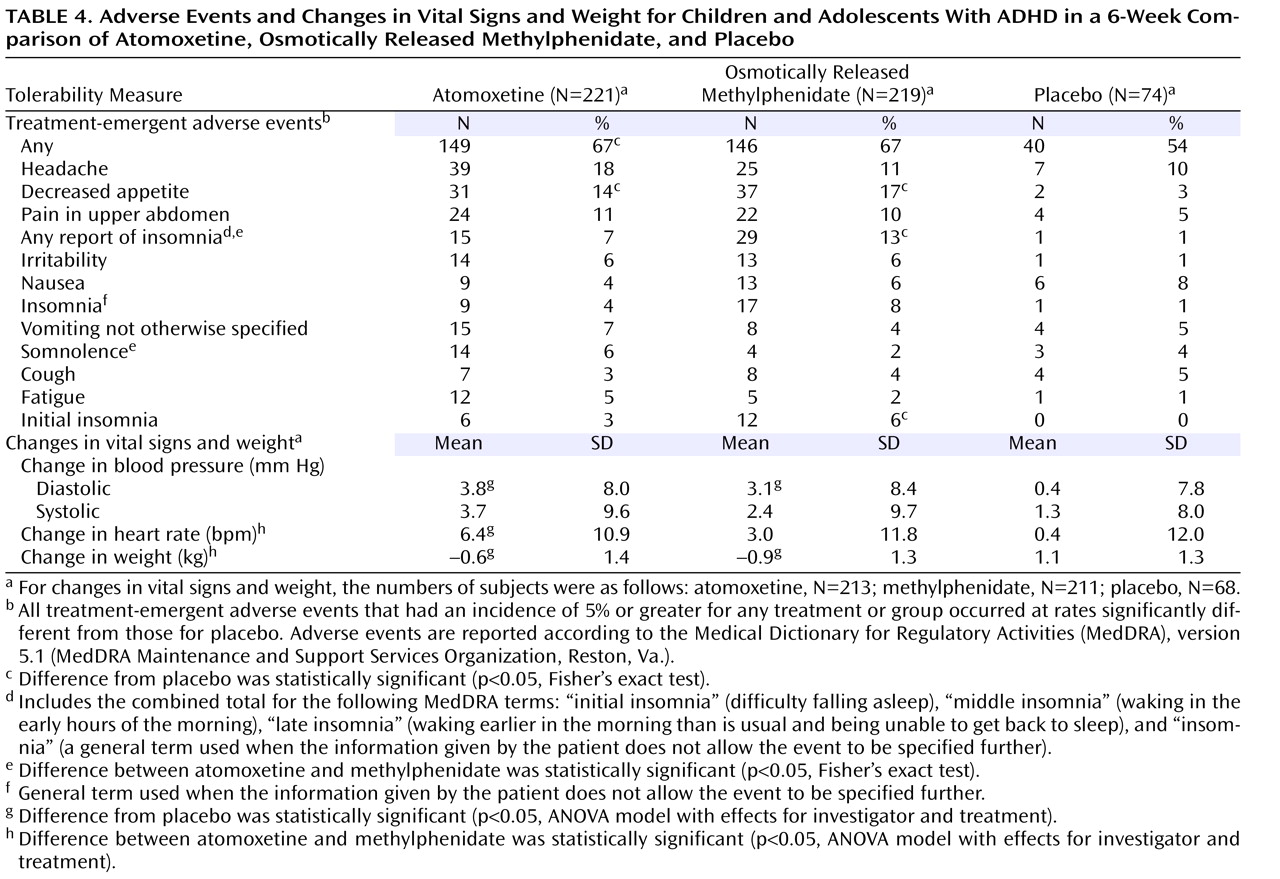
Adverse events that occurred in at least 5% of the patients in any treatment group or that occurred significantly more often for either drug than for placebo are listed in
Table 4 . The only event that was significantly different from placebo for both drugs was decreased appetite. Insomnia (“any report of insomnia”) was more common for patients assigned to methylphenidate than for those taking placebo. Somnolence was reported more often for atomoxetine than for methylphenidate, while insomnia was reported more often for methylphenidate than for atomoxetine.
Changes in weight and vital sign measurements are also presented in
Table 4 . The mean increase in diastolic blood pressure, relative to placebo, was statistically significant for both atomoxetine and osmotically released methylphenidate. No differences were observed in mean change of systolic blood pressure between placebo and either drug. Increase in heart rate was significantly greater for atomoxetine than for either placebo or methylphenidate. Weight loss was significantly greater with both atomoxetine and methylphenidate than with placebo, and the magnitude of weight loss was greater for methylphenidate than for atomoxetine. Neither drug was associated with meaningful changes in laboratory measures.
Assessment 2: Crossover From Methylphenidate to Atomoxetine
Of the 220 patients who were randomly assigned to receive osmotically released methylphenidate, 180 completed 6 weeks of treatment. Forty subjects (18%) originally assigned to methylphenidate dropped out during the acute treatment phase and therefore were not eligible to receive crossover treatment. Of the remaining subjects, 178 (99%) were switched to atomoxetine. The responses to the two treatments in these subjects were as follows: 60 of the 178 (34%) responded to either atomoxetine or osmotically released methylphenidate but not both, 78 (44%) responded to both treatments, and 40 (22%) did not respond to either treatment. Of the 70 patients who did not respond to methylphenidate in the initial trial, 30 (43%) subsequently responded to atomoxetine. Of the 69 patients who did not respond to atomoxetine in the second trial, 29 (42%) had previously responded to osmotically released methylphenidate. Similar results were obtained when these analyses were restricted to patients who received at least 1 week of atomoxetine treatment (methylphenidate responders, N=100; methylphenidate nonresponders, N=64).
Additional post hoc analyses were conducted to examine the possibility that using a categorical response criterion masked clinically significant improvement that nevertheless fell short of the specified threshold, as well as the possibility that reaching or falling below the threshold for positive response represented only a modest difference in response between the two medications. For all patients originally treated with osmotically released methylphenidate and subsequently switched to atomoxetine, we calculated the percentage of improvement (relative to the baseline value) seen at the end of the acute treatment comparison and at the last available visit in the crossover trial. We then classified each of these patients as having 1) a “significantly better response” if the improvement with atomoxetine at assessment 2 was greater than or equal to the improvement with methylphenidate at assessment 1, plus 15%, 2) “not significantly different response” if the improvement with atomoxetine at assessment 2 was within 15% of the improvement at assessment 1, or 3) “significantly worse response” if the improvement with atomoxetine at assessment 2 was less than or equal to the assessment 1 improvement, minus 15%. We set the threshold for this analysis at 15% to represent the difference between an accepted, liberal threshold of response (25% reduction in ADHD symptoms) and the more stringent 40% threshold we used to define response in this study.
Of the 108 patients classified as methylphenidate responders, 39 (36%) showed significantly worse response, 19 (18%) showed significantly better response, and 50 (46%) showed roughly the same response to treatment with atomoxetine. Of the 70 patients classified as methylphenidate nonresponders, seven (10%) showed significantly worse response, 36 (51%) showed significantly better response, and 27 (39%) showed roughly the same response to treatment with atomoxetine. Similar results were obtained when these analyses were performed for only the patients who received at least 1 week of atomoxetine treatment.
Similar, high proportions of methylphenidate nonresponders (70%) and methylphenidate responders (81%) completed the second trial. Discontinuations as a result of adverse events were infrequent and similar for both groups. We are not able to report on either response to or tolerability of atomoxetine in subjects who terminated participation in the initial comparison, since these subjects did not have an opportunity to receive treatment with atomoxetine. However, in that assessment the dropout rate due to poor response or adverse events with methylphenidate (N=9) was low, and the majority of subjects dropped out for other reasons (N=31).
Discussion
There has been considerable speculation about the relative efficacy and tolerability of atomoxetine compared with stimulant treatments for ADHD. To our knowledge, this is the first large double-blind, randomized, placebo-controlled study to prospectively compare response to atomoxetine and methylphenidate in this population. Additionally, the study provides preliminary information regarding differential response to the two treatments. Both atomoxetine and osmotically released methylphenidate were superior to placebo, with moderate to large effects for each treatment. Response to osmotically released methylphenidate was superior to that for atomoxetine. There was some evidence for differential response, with one-third of the subjects responding to either methylphenidate or atomoxetine but not both.
Interpretations of comparator trials are inherently limited by design features that might favor one or the other treatment, and several factors in the present study could have influenced the observed results. Subjects who previously had either poor response or intolerable adverse events in an adequate trial of methylphenidate or any other stimulant were excluded for ethical reasons, a criterion that was not possible for atomoxetine because it was not commercially available at the time the study was undertaken. Patients with comorbid tics and anxiety disorders were likewise excluded, because these are labeled contraindications to methylphenidate. Either of these criteria could have created a bias in favor of methylphenidate by excluding subjects with increased risk for poor response or tolerability with methylphenidate.
Conversely, design features related to dose may have created a bias in favor of atomoxetine. Atomoxetine was titrated up to a maximum dose of 1.8 mg/kg on the basis of previous experiences in pivotal efficacy trials. This dose is higher than the maximum dose of atomoxetine (1.4 mg/kg) subsequently approved by the Food and Drug Administration (FDA), which was derived from findings in a dose-ranging study
(2), in which response to atomoxetine was not greater in subjects receiving 1.8 mg/kg per day than in those receiving 1.2 mg/kg per day. In contrast, the maximum dose of osmotically released methylphenidate was 54 mg/day, which was the maximum labeled dose for this medication at the time the study was conducted but is lower than the current labeled maximum of 72 mg/day for adolescents. In this study, adolescents (N=57) received somewhat lower weight-based doses of osmotically released methylphenidate than children (N=162) (0.88 mg/kg versus 1.26 mg/kg). Of note, the mean methylphenidate dose for adolescents in this study was virtually identical to that in a recent controlled study of osmotically released methylphenidate in adolescents
(24), and it is only somewhat lower than in the community-based comparator trial of osmotically released methylphenidate and atomoxetine reported by Kemner et al.
(8) . However, it is likely that the methylphenidate dose was suboptimal for some adolescent subjects. Restricting the dose of methylphenidate to 54 mg could also have limited response in some younger subjects, since osmotically released methylphenidate is sometimes prescribed at doses higher than the FDA-recommended maximum for children
(11) . However, it is noteworthy that the dose
range for osmotically released methylphenidate in this study was quite large (i.e., up to 2.57 mg/kg per day), which suggests that capping the dose of osmotically released methylphenidate at 54 mg was likely not problematic for many of the younger children.
Another design decision that could have altered the study findings was the administration of atomoxetine as a twice-daily divided dose. Trials using once- and twice-daily dosing of atomoxetine have produced similar results
(3), but we cannot rule out the possibility that twice-daily dosing favored atomoxetine and that once-daily administration might have yielded poorer efficacy or tolerability. Also, there were no direct reports from teachers. Available meta-analyses indicate that parent reports are at least as sensitive as teacher reports at detecting change in overall ADHD symptoms as measured on standard instruments such as the ADHD Rating Scale
(25 –
27) . However, if the two medications had differential effects on ADHD symptoms or behavior during the school day, this might not have been detected. Finally, the stimulant comparator in this study was osmotically released methylphenidate; findings may not generalize to other formulations of methylphenidate or to amphetamine.
The preceding potential limitations notwithstanding, there are several important clinical implications of this study. The magnitude of response to atomoxetine is presented in the context of a frequently used, long-acting stimulant comparator. While atomoxetine did not perform as well as osmotically released methylphenidate overall, response to atomoxetine was nevertheless solid, with an effect size within 0.2 of that achieved for methylphenidate. Including both effect size and number needed to treat in the data analysis offers two ways to evaluate the overall differences in treatment efficacy.
Another important finding relates to the group of patients switched to atomoxetine after completing 6 weeks of methylphenidate. About one-half of these subjects responded robustly to both treatments. However, approximately two-thirds of the others responded preferentially to one treatment, divided approximately equally between methylphenidate and atomoxetine. Further, a smaller but not insignificant number (22%) were nonresponders to both treatments. This finding is consistent with the results of a previous smaller, order-randomized crossover study of immediate-release methylphenidate and atomoxetine
(28), and it suggests that subgroups of patients may derive greater benefit from one or the other treatment. This may be attributable to differential sensitivity to the pharmacologic mechanisms, individual metabolic and pharmacokinetic responses, or other reasons.
It is noteworthy that the crossover study was conducted under double-blind conditions. However, there were several other limitations in design. Most important, order effects could have contributed to the differential response findings, since subjects who were crossed over always received methylphenidate first and atomoxetine second. In addition, 18% of the subjects originally randomly assigned to osmotically released methylphenidate discontinued the study before entering the crossover period, and therefore they did not receive atomoxetine. Finally, there was no washout period between the two treatment phases. Although it is unlikely that carryover effects of osmotically released methylphenidate would persist after 6 weeks of treatment with atomoxetine, we cannot definitively rule out this possibility. Because of the preceding limitations, the results of these crossover analyses must be considered preliminary. They are nonetheless presented here because no other large-scale comparator data of this sort currently exist and because we know of no other study that is likely to generate similar data for several years. Most important, the finding that there are subgroups of patients who respond preferentially to osmotically released methylphenidate or atomoxetine is of considerable clinical importance and provides at least some empirical support for clinical guidelines that recommend using a second medication class if treatment with the first agent is unsatisfactory
(11) .
Overall, both atomoxetine and osmotically released methylphenidate were safe and well tolerated, with only modest differences in tolerability and vital signs between the two drugs. Both medications had more reports of decreased appetite than placebo. Atomoxetine was more likely to be accompanied by somnolence, while methylphenidate was more often associated with insomnia. Both drugs were associated with modest increases in cardiovascular tone, with greater heart rate increases in the atomoxetine patients than the methylphenidate patients—perhaps reflecting the twice-daily dosing schedule for atomoxetine. Both drugs were also associated with greater weight loss than placebo, an effect that was greater for methylphenidate than atomoxetine. Neither drug was associated with treatment-emergent suicidal ideation, although concerns about the potential for increased suicidality related to medications used to treat ADHD have been raised
(29) and have recently been added to the product labeling for atomoxetine. However, it should be noted that the number of subjects was based primarily on detecting differences in efficacy, not safety, and it is possible that infrequently occurring events could have been missed. This limitation notwithstanding, the group tolerability data do not seem to favor one treatment over the other, with individual differences likely being more important in guiding clinical use.
In summary, atomoxetine and osmotically released methylphenidate were evaluated in a large placebo-controlled, randomized, double-blind, parallel-group study. Both treatments produced robust improvement, with a statistically significant difference in response favoring osmotically released methylphenidate. Data from a subsequent crossover from methylphenidate to atomoxetine provided evidence for differential response to the two treatments in approximately one-third of the patients. This finding is consistent with practice guidelines that recommend change to a different class of medication if there is poor response to or tolerance of the first agent used.