This analysis compared participants who would meet typical inclusion and exclusion criteria used in phase III trials (efficacy sample) with those who would not (nonefficacy sample) with regard to baseline sociodemographic and clinical characteristics, treatment characteristics, and treatment outcomes (as measured by depressive symptoms and adverse events). To our knowledge, this study is the first to examine differences in treatment outcomes between efficacy and nonefficacy samples.
Method
Study Overview and Organization
The rationale and design of STAR*D are detailed elsewhere
(2,
3) . The purpose of STAR*D was to define prospectively which of several treatments are most effective for outpatients with nonpsychotic major depressive disorder who have an unsatisfactory clinical outcome to an initial and, if necessary, subsequent treatment(s). Between July 2001 and April 2004, STAR*D enrolled participants at 18 primary care and 23 psychiatric specialty care settings across the United States.
Study Sample
The study protocol was approved and monitored by the institutional review boards at the national coordinating center (Dallas), the data coordinating center (Pittsburgh), each clinical site and regional center, and the data safety and monitoring board of the National Institute of Mental Health (NIMH) (Bethesda, Md.). All risks and benefits associated with STAR*D participation were explained to the participants, who provided written informed consent before study entry.
To enhance the generalizability of the results, only self-declared outpatients seeking treatment in either primary care or specialty care settings and identified by their clinicians as having major depressive disorder that required treatment were eligible. Advertising for symptomatic volunteers was proscribed. Broadly inclusive selection criteria were used to ensure recruitment of a representative sample. Eligible participants were 18–75 years of age, met the DSM-IV criteria for single-episode or recurrent nonpsychotic major depressive disorder (established by treating clinicians and confirmed by a DSM-IV checklist), scored 14 or higher (moderate severity) on the 17-item version of the Hamilton Depression Rating Scale (HAM-D)
(5,
6) (rated by the clinical research coordinators at each site), and had not been found to be treatment resistant in an adequate antidepressant trial during the current major depressive episode. Patients were excluded if they were pregnant, intending to become pregnant, or breastfeeding; had a primary psychiatric disorder requiring a different treatment approach (a bipolar, psychotic, obsessive-compulsive, or eating disorder); had substance abuse or dependence that required inpatient detoxification; were using medications excluded by the study; or had a seizure disorder or other general medical condition that contraindicated medications used in the first two protocol treatment steps. All other psychiatric and medical comorbidities were allowed.
Baseline Measures
At baseline, the clinical research coordinators collected standard sociodemographic information, self-reported psychiatric history, and information on current general medical conditions as evaluated by the Cumulative Illness Rating Scale
(7,
8) . In addition to administering the initial HAM-D, the clinical research coordinators assessed depressive symptom severity using the 16-item Quick Inventory of Depressive Symptomatology—Clinician-Rated, and the participant completed the Quick Inventory of Depressive Symptomatology—Self-Report
(9 –
12) . Participants also completed the Psychiatric Diagnostic Screening Questionnaire
(13,
14), which was used to estimate the presence of 11 potential concurrent DSM-IV disorders.
The research outcomes assessors, blinded to treatment and not located at any site, used a telephone interview at baseline to administer the HAM-D and the 30-item clinician-rated Inventory of Depressive Symptomatology
(9,
12,
15) to measure core symptoms and associated symptoms of depression. Responses to items on these measures were used to estimate the presence of atypical
(16), anxious
(17), and melancholic
(18) symptom features.
Intervention
Citalopram was selected as a representative SSRI given the relative absence of discontinuation symptoms, demonstrated safety in elderly and medically fragile patients, once-a-day dosing, few dose-adjustment steps, anticipated generic availability, and favorable drug-drug interaction profile
(19) . The aim of treatment was to achieve symptom remission (defined as a score of 5 or less on the self-rated Quick Inventory of Depressive Symptomatology, which was administered at each treatment visit for the purposes of clinical decision making). The protocol required a fully adequate dose of citalopram for a sufficient time to ensure that the likelihood of reaching remission was maximized and that participants who did not reach remission were truly experiencing inadequate benefit from the medication.
The protocol aimed to provide an optimal dose of citalopram based on dosing recommendations in a treatment manual (www.star-d.org). Citalopram was to be started at 20 mg/day, then raised to 40 mg/day by week 4, and raised to the final dose of 60 mg/day by week 6. Dose adjustments were guided by symptom changes (Quick Inventory of Depressive Symptomatology completed by the clinical research coordinator), side effect burden (according to the Frequency, Intensity, and Burden of Side Effects Rating [FIBSER]
[20] ), and how long a participant had received a particular dose. The protocol guided physicians to make management decisions at weeks 4, 6, 9, and 12. These were critical decision points at which a decision could be made to modify the dose and/or address side effects or to move to the next treatment level. Still, appropriate flexibility was allowed to minimize side effects, maximize safety, and optimize the chances of therapeutic benefit for each participant. This included initiation of citalopram at a dose below 20 mg/day or a slower dose escalation to the optimal target dose of 60 mg/day. In this way, the study could safely include patients with concomitant general medical disorders, substance abuse or dependence, or other psychiatric disorders and those sensitive to medication side effects.
The protocol recommended treatment visits at weeks 2, 4, 6, 9, and 12 (with an optional week 14 visit if needed). After an optimal trial (as judged by dose and duration), patients with remission could enter the 12-month naturalistic follow-up, as could responders without remission, although all of those without remission were encouraged to enter the subsequent randomized trial (level 2 of STAR*D). Participants could discontinue citalopram before 12 weeks if 1) intolerable side effects required a medication change, 2) an optimal dose increase was not possible because of side effects or participant choice, or 3) significant symptoms (score of 9 or higher on the clinician-rated Quick Inventory of Depressive Symptomatology) were present after 9 weeks at the maximally tolerated dose. Participants could opt to move to the next treatment level if they had intolerable side effects or if the score on the clinician-rated Quick Inventory of Depressive Symptomatology was higher than 5 after an adequate trial in terms of dose and duration
(4) .
Intensive efforts to provide consistent, high-quality care are represented by the use of a treatment manual, initial didactic instruction, ongoing support and guidance by the clinical research coordinators, the use of structured evaluation of depressive symptoms and side effects at each visit, and a centralized treatment monitoring and feedback system (www.star-d.org) that provided feedback to clinical research coordinators regarding each participant’s fidelity to the treatment recommendations. The clinical research coordinators could then help guide physicians in vigorous dosing when inadequate symptom reduction had occurred despite acceptable side effects
(4) .
Safety Assessments
In addition to side effects, serious adverse events were monitored with a multitiered approach involving the clinical research coordinators, study clinicians, interactive voice response system, safety officers, regional center directors, and NIMH data safety and monitoring board
(3) .
Concomitant Medications
Concomitant treatments for current general medical conditions (as part of ongoing clinical care), for associated symptoms of depression (e.g., sleep, anxiety, and agitation), and for citalopram side effects (e.g., sexual dysfunction) were permitted on the basis of clinical judgment. The protocol prohibited the use of stimulants, anticonvulsants, antipsychotics, alprazolam, nonprotocol antidepressants (except trazodone at a dose of 200 mg or less at bedtime for insomnia), and depression-targeted psychotherapies.
Primary Outcome Measures
Phase III trials traditionally assess outcomes 8 weeks after random assignment of treatment. In STAR*D, clinic visits were scheduled at 2, 4, 6, 9, and 12 weeks after enrollment. The week 9 assessment was used to approximate the time frame of the phase III trial. The primary outcome was based on the self-rated Quick Inventory of Depressive Symptomatology, which was administered at baseline and at each treatment visit. Remission was defined as a score of 5 or less (which is equivalent to a score of 7 or less on the 17-item HAM-D)
(11) at week 9 or, if the last visit occurred before week 9, the last recorded score. The secondary outcome was response, which was defined as a reduction of at least 50% from the baseline score on the self-rated Quick Inventory of Depressive Symptomatology at the last assessment at or before week 9.
Defining Efficacy and Nonefficacy Samples
The whole of the STAR*D sample was consistent with a study group enrolled in an effectiveness trial. The criteria for inclusion in the efficacy sample were established a priori by consensus of several authors (A.J.R., M.H.T., M.F., A.A.N., P.J.M., B.N.G.) on the basis of their experience in designing and implementing placebo-controlled registration trials. The efficacy sample met all of the following criteria: 1) baseline HAM-D score higher than 19 (assessed by the clinical research coordinator), 2) no more than one concurrent general medical condition (defined as no more than one item of the Cumulative Illness Rating Scale with a score higher than 1), 3) the absence of obsessive-compulsive disorder (according to the Psychiatric Diagnostic Screening Questionnaire), 4) no more than one additional concurrent axis I psychiatric disorder (according to the Psychiatric Diagnostic Screening Questionnaire), and 5) a current episode lasting less than 24 months.
Those who did not meet the criteria for inclusion in the efficacy sample were included in the nonefficacy sample.
Statistical Analysis
Summary statistics are presented as means and standard deviations for continuous variables and as percentages for discrete variables. Student’s t tests and Mann-Whitney U tests were used to compare continuous baseline sociodemographic and clinical features, treatment features, side effect rates, and rates of serious adverse events in the two samples. Chi-square tests were used to compare discrete characteristics in the two samples.
Logistic regression models were used to compare remission and response rates, after adjustment for the effect of baseline characteristics that were not equally distributed across the two groups. Times to first remission and first response were defined as the first observed point in the clinic visit data. Log-rank tests were used to compare the cumulative proportions of participants in each sample who reached remission or response. Additional exploratory logistic regression analyses were conducted to determine if there was a differential (moderating) effect of treatment setting (psychiatric care or primary care) on remission based on the severity of depression, as judged by the baseline score on the self-rated Quick Inventory of Depressive Symptomatology.
Statistical significance was defined as a two-sided p value of <0.05. No adjustments were made for multiple comparisons, so the results must be interpreted accordingly.
Results
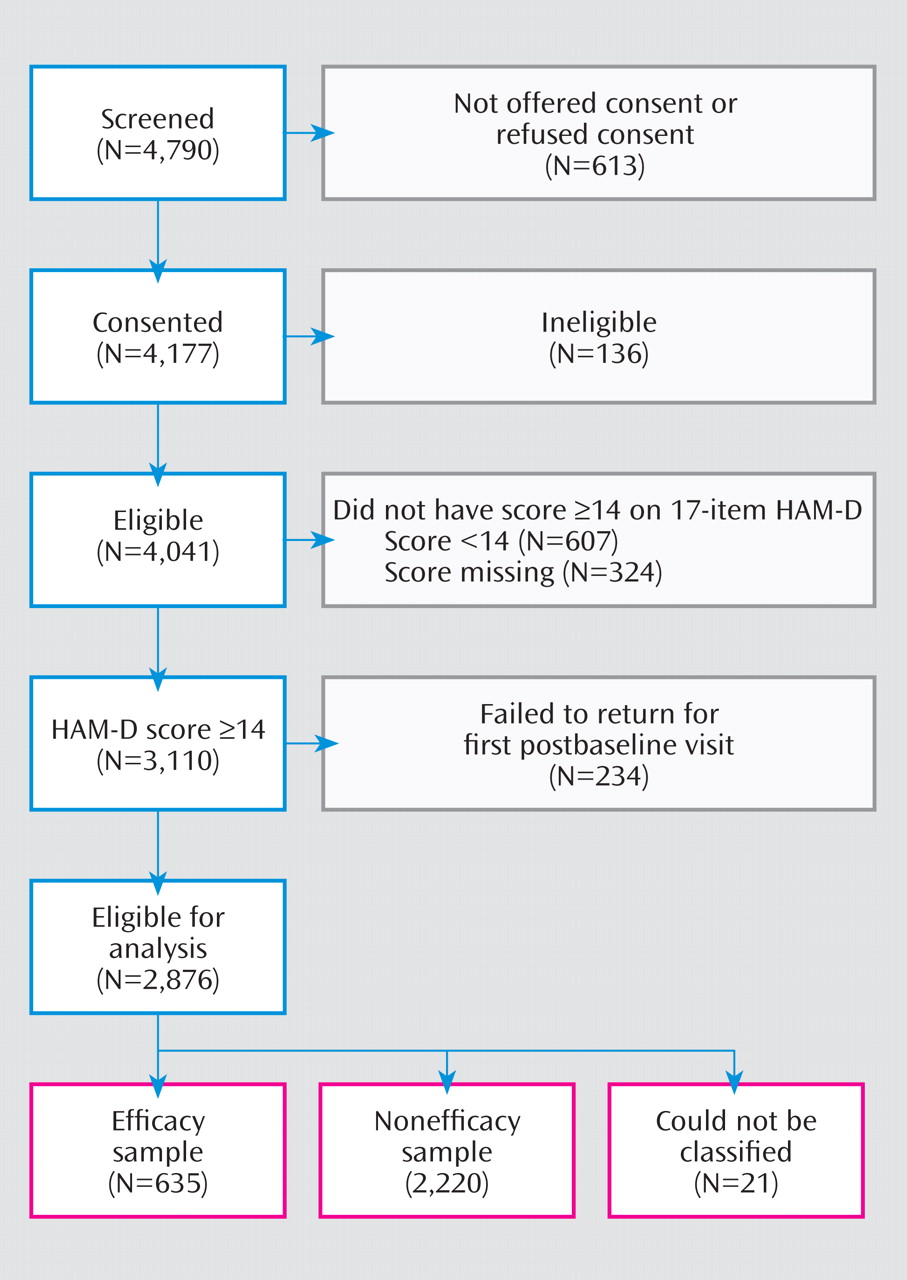
STAR*D enrolled a total of 4,041 participants, 2,876 of whom made up an analyzable sample (having at least one postbaseline visit and a score of 14 or higher on the HAM-D). Of these, 2,855 could be classified into the efficacy sample (N=635, 22.2%) or the nonefficacy sample (N=2,220, 77.8%) (
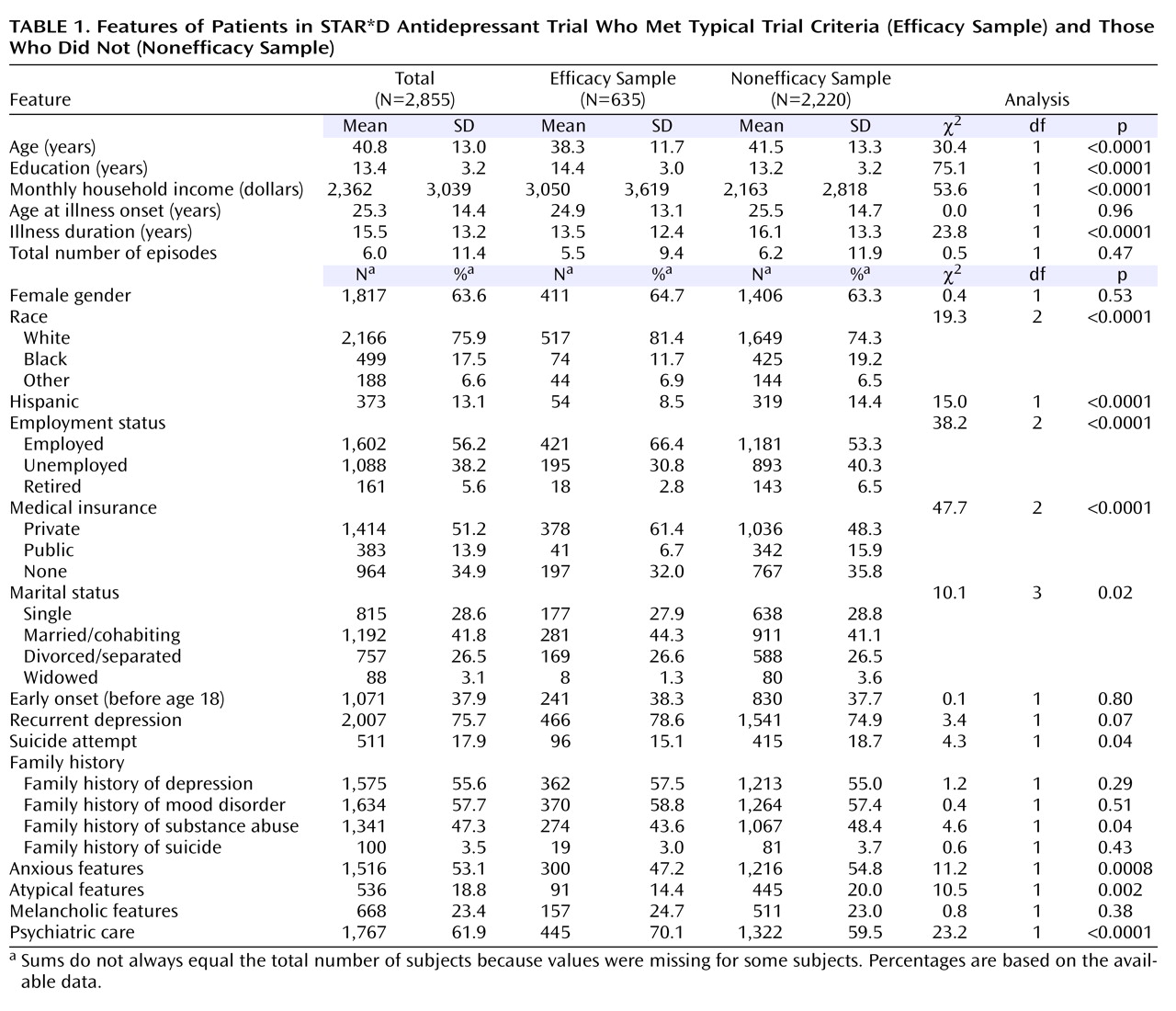
Figure 1 ). On average, participants in the efficacy sample were more likely to be younger, more educated, white, non-Hispanic, employed, married, and privately insured and to have a higher income (
Table 1 ). The efficacy group also had a shorter average duration of illness (time from onset of the first episode of major depressive disorder to study enrollment) and lower rates of prior suicide attempts, family history of substance abuse, and anxious or atypical symptom features. More participants in the efficacy sample were seen in psychiatric specialty care settings.
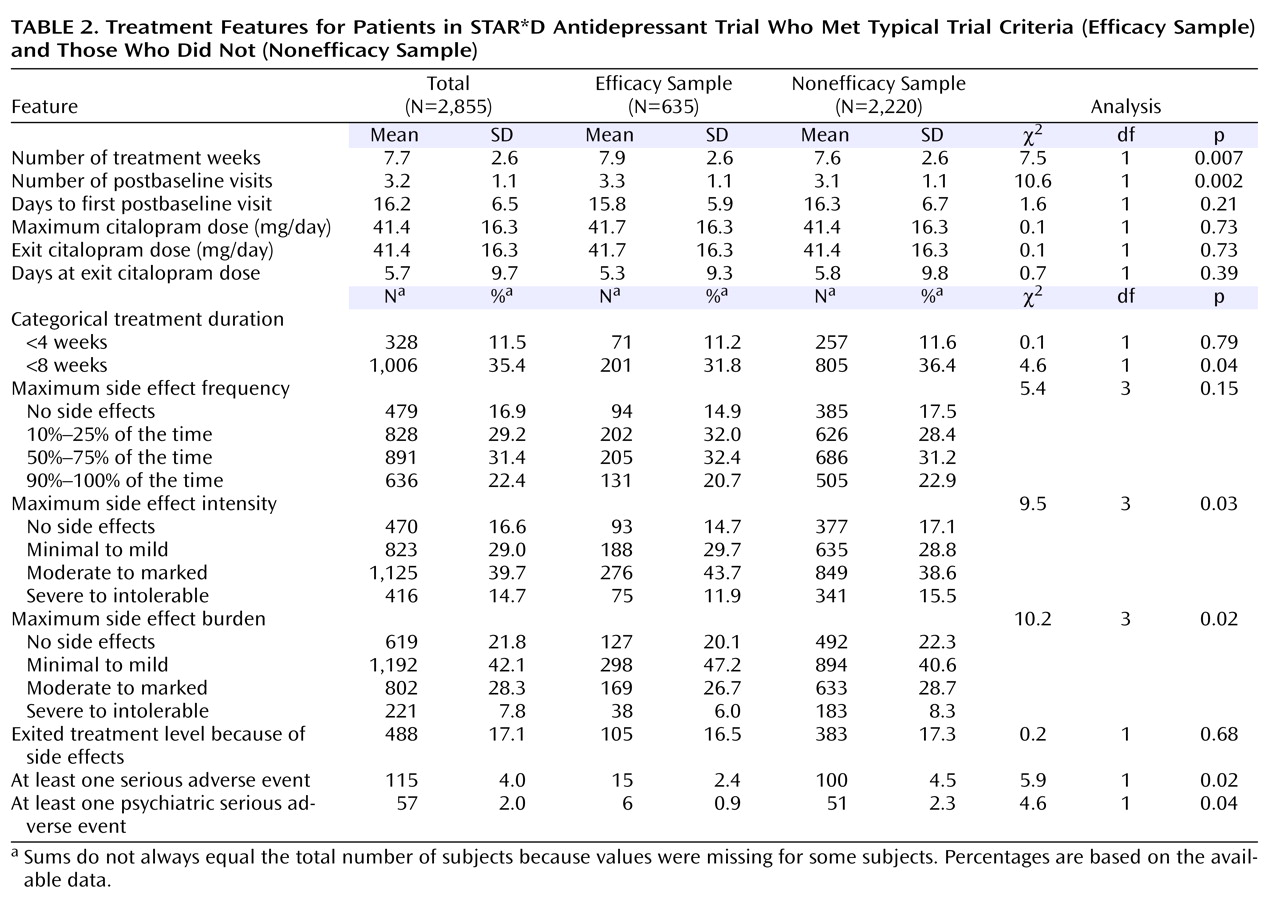
Participants in the efficacy sample were less likely to have side effects of severe or intolerable intensity, moderate to intolerable side effect burden, serious adverse events, and psychiatric serious adverse events (
Table 2 ). Of note, there were no significant differences between groups in the dosing of citalopram (maximum dose or exit dose) or in the number of days at the exit dose. Participants in the efficacy sample had, on average, more weeks in treatment and more clinic visits, although these differences were not clinically meaningful.
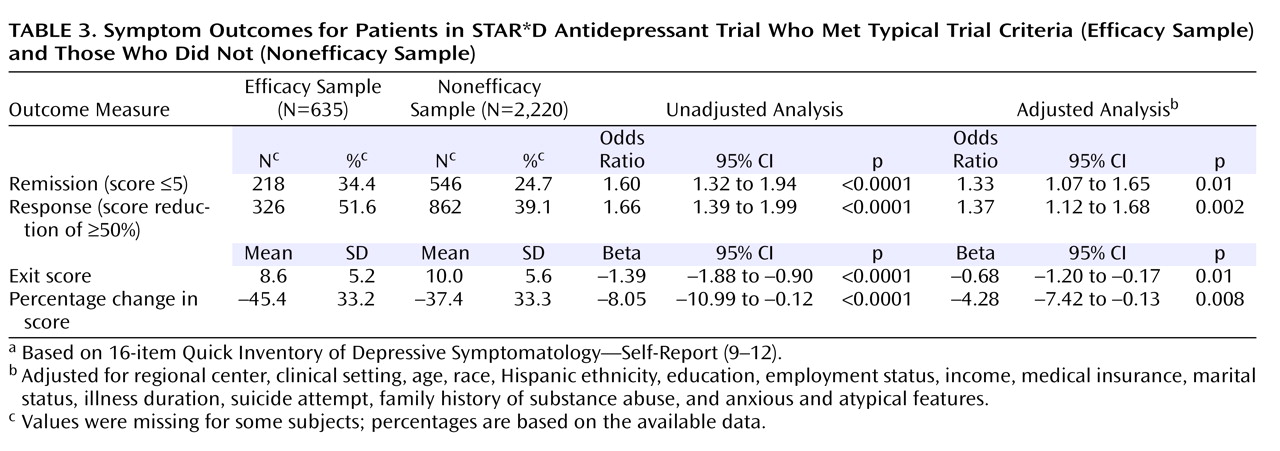
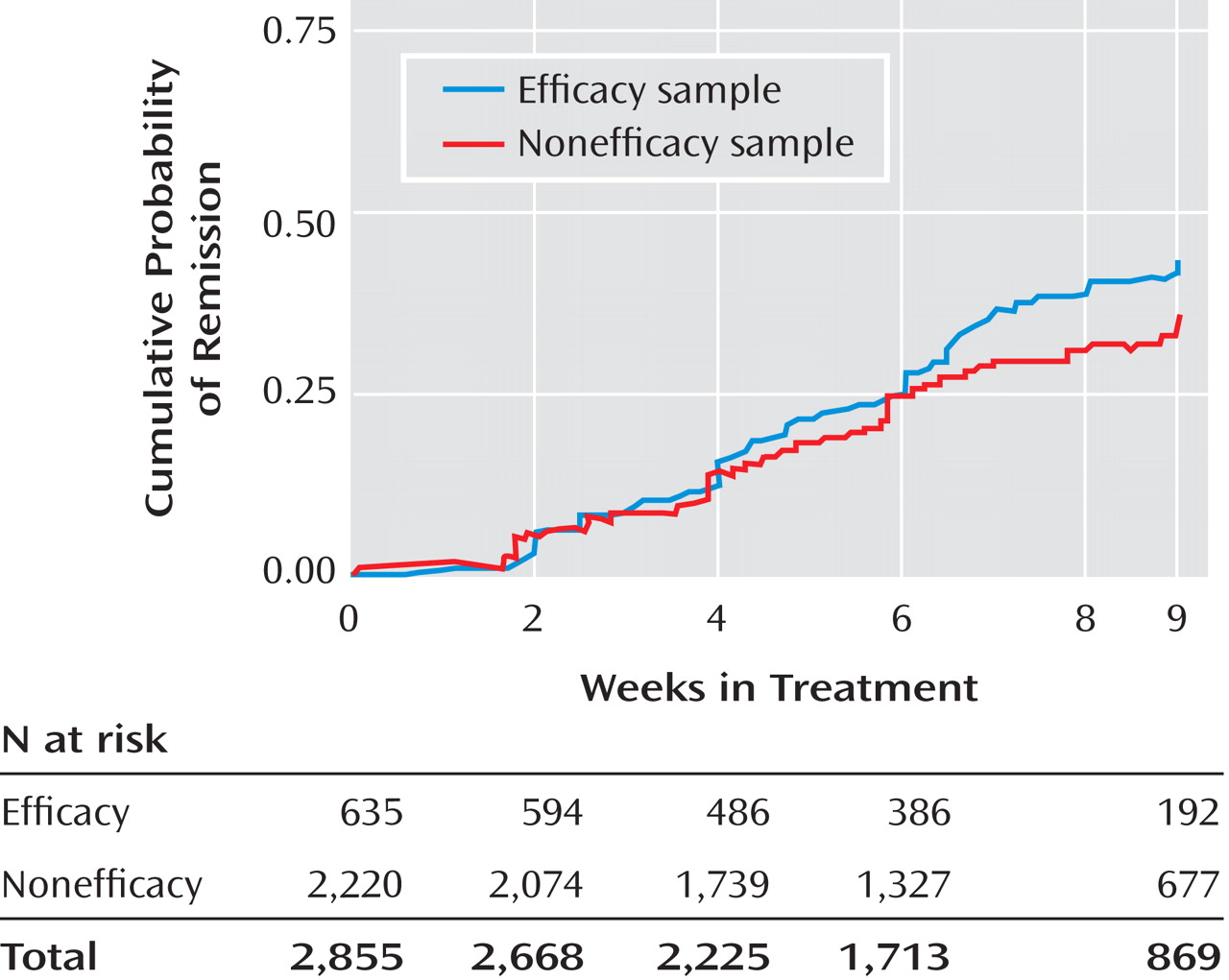
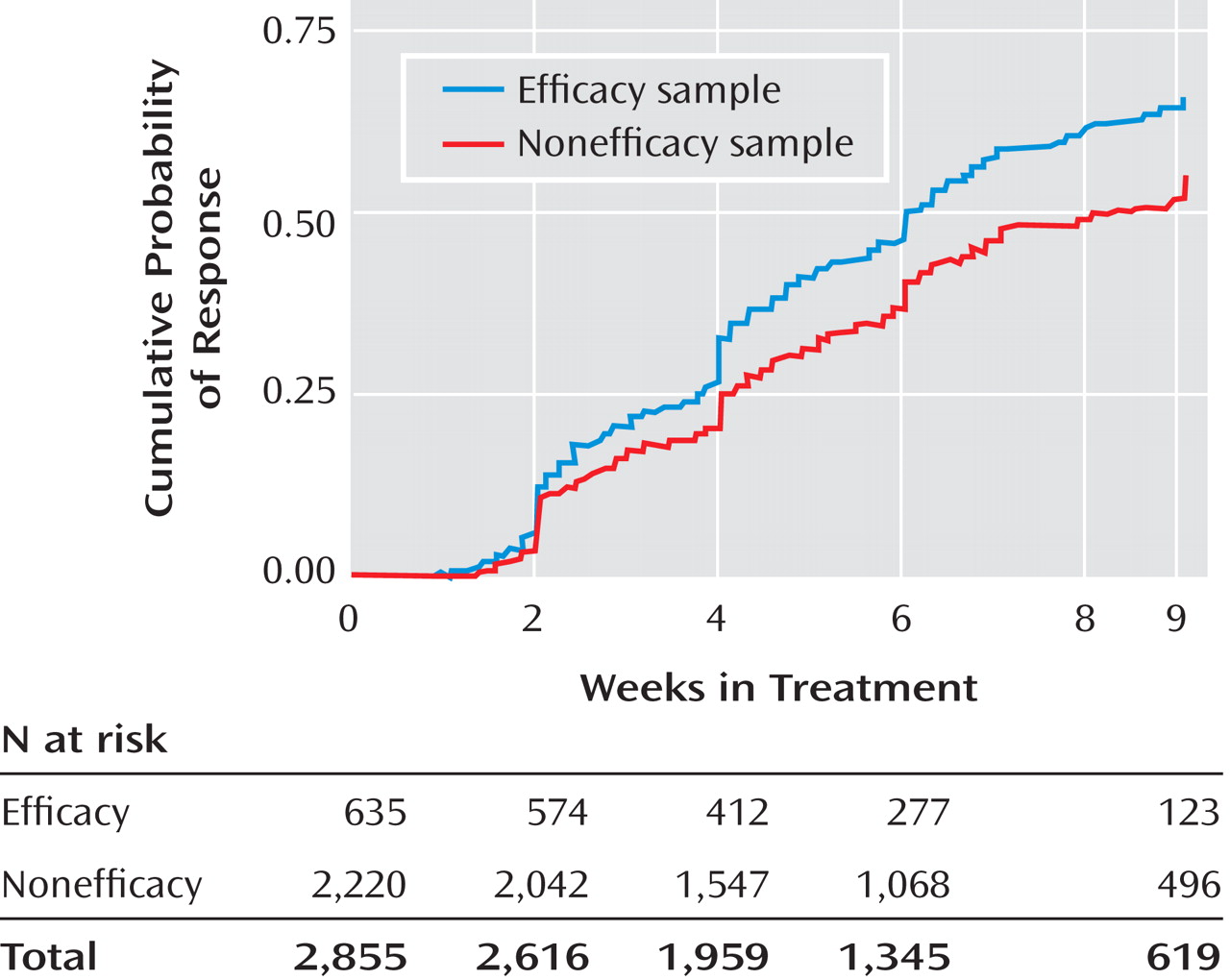
The remission rates were 34.4% in the efficacy sample and 24.7% in the nonefficacy sample, and the number needed to treat was 10. The response rate was also lower in the nonefficacy group (51.6% versus 39.1%). Even after adjustment for potential baseline confounding characteristics, the efficacy sample had significantly better depression symptom outcomes (
Table 3 ). They also had a shorter time to remission (
Figure 2 ) and time to response (
Figure 3 ). For those who achieved response, the mean time to response was 4.6 weeks (SD=2.4) for the efficacy sample and 4.8 weeks (SD=2.5) for the nonefficacy sample. For those who achieved remission, the mean time to remission was 5.5 weeks (SD=2.5) for the efficacy sample and 5.3 weeks (SD=2.5) for the nonefficacy sample. Serious adverse events were classified by the type of event. The two most prevalent events were psychiatric hospitalizations and general medical hospitalizations. The groups differed in the rate of psychiatric hospitalizations; for the efficacy sample the percentage was 0.3% (two of 635), and for the nonefficacy sample it was 2.5% (56 of 2,220) (χ
2 =12.1, df=1, p<0.001). They also differed in the rate of general medical hospitalization; for the efficacy sample the rate was 1.1% (seven of 635, and for the nonefficacy sample it was 2.7% (60 of 2,220) (χ
2 =5.1, df=1, p=0.02).
Discussion
Fewer than one in four (22.2%) of the participants met the criteria for inclusion in the efficacy sample. Such a finding in a group as large and generalizable as the STAR*D sample indicates that a comparably small percentage of depressed patients treated in primary and psychiatric care settings would meet these criteria. Therefore, since the efficacy sample was based on phase III clinical trial criteria, it seems that these criteria would similarly recruit only a small percentage of typical depressed patients into phase III trials.
We found numerous differences in baseline sociodemographic and clinical characteristics between the efficacy and nonefficacy samples and a few differences regarding treatment characteristics. The latter were mostly related to side effects, although both groups received relatively equivalent doses of citalopram. Further, all measures of outcome showed significant but modest differences between the groups, with the efficacy sample having, on average, better outcomes. These differences were consistent in the direction and magnitude of effect when examined separately in primary and psychiatric care settings.
Given these between-group differences, the smaller efficacy sample is clearly not representative of the more inclusive, treatment-seeking population. By inference, a patient sample that meets the inclusion criteria for a phase III clinical trial is not representative of depressed patients seen in typical clinical practice, and phase III trial outcomes may be more optimistic than results obtained in practice.
The issue of the generalizability of randomized clinical trials is a topic that is discussed in the medical literature
(21) . The concern is that the results of randomized clinical trials are often poorly generalizable to a real-world clinic population, which could lead to the underuse of effective treatments or the overuse of ineffective treatments. This concern arises in both general medicine and psychiatry. Regarding general medicine, Fortin et al.
(22) found that in randomized clinical trials targeting a chronic medical condition, most eligible patients had comorbid conditions that precluded eligibility. In psychiatry, Zimmerman et al.
(23) found that of 315 patients with major depressive disorder who sought care, only 29 (9.2%) met typical inclusion and exclusion criteria for an efficacy trial. Kessler et al.
(24) noted that most real-world patients with major depression would be excluded from randomized, controlled trials because of comorbid conditions. This existing literature, along with our study, highlights the broad public health value of large practical clinical trials and provides a model for how evidence-based psychiatry may be introduced into real-world clinics.
Thus, our results are largely consistent with previous findings that outpatients included in phase III randomized, controlled efficacy trials for major depressive disorder are different from those who would be excluded. These previous studies, however, have only examined baseline characteristics.
To our knowledge, the current study is the first to examine the differences in treatment outcome. Notably, response and remission rates were poorer and the times to response and remission were longer in patients ineligible for efficacy trials. Thus, current efficacy trials suggest a more optimistic outcome than is likely in practice, and the duration of adequate treatment suggested by data from efficacy trials may be too short.
Our findings could have significant implications for the future design of phase III trials for antidepressant treatment. Perhaps the inclusion criteria for phase III trials could be expanded to generate more generalizable information on the safety and efficacy of antidepressants, but this could come at the cost of a somewhat greater risk of adverse events. The traditional phase III approach assesses treatment efficacy in only a small subset of the population for which the treatment is intended. Therefore, a treatment defined as efficacious in the relatively small study group may be less effective and perhaps not as well tolerated in larger populations. To adequately assess whether this is so, one would have to determine if the efficacy sample has a differential treatment response in a placebo-controlled trial, which is not possible in the current study given the STAR*D design.
In addition, placebo response rates and detectable effect sizes in phase III trials might be reduced by recruiting more representative participants, including patients with concurrent comorbidity and other features (e.g., chronicity), which would increase the efficiency while improving the generalizability of phase III trials. Several studies have found differences among these populations and reduced placebo responsiveness in the presence of such features
(25 –
27) .
The present study has several limitations. First, there are no standard inclusion and exclusion criteria for a phase III clinical trial. The characteristics we used to define the efficacy sample in this study were based on an approximation of what is commonly used for a phase III clinical trial. The sensitivity of the current study’s criteria was assessed by varying the assumptions to re-create the efficacy and nonefficacy samples by using other criteria and repeating the analyses. Specifically, a more stringent criterion was used that required no prior history of a suicide attempt and no current risk of suicide in addition to the earlier stated criteria for the efficacy sample. As a result of the modification, the size of the efficacy sample decreased from 635 to 522. The association of the sample with outcome remained relatively unchanged. For example, the unadjusted odds ratio for remission changed from 1.60 in the original analysis to 1.64 in the sensitivity analysis, while the adjusted odds ratio changed from 1.33 to 1.26. Thus, the conclusions derived from the sensitivity sample are identical to those derived from the original analyses.
Another limitation is the use of self-report rather than clinical interviews to assess psychiatric and general medical comorbidities. While this limits the comparability to phase III trials, it does help in generalizing findings to standard clinic practice, where clinicians tend not to use diagnostic instruments (e.g., Structured Clinical Interview for DSM-IV) but instead use self-report. Further, the efficacy sample developed for this study is not fully representative of phase III clinical samples because, unlike most phase III trials, STAR*D proscribed the enrollment of symptomatic participants recruited by advertising. It is likely that the differences in outcomes for the two study groups would be even more pronounced for a phase III trial consisting of participants who are typical symptomatic volunteers. Also, STAR*D’s broader inclusion criteria were justified by the enormous safety data available for citalopram, which was administered open-label. Most pivotal clinical trials for registration test compounds that have far less safety information and essentially no information regarding their effect on comorbid medical conditions. In the case of investigational compounds, the lack of demonstrated efficacy and the exiguous safety information would make broad inclusion less justified. The lack of placebo and double-blinding may also affect treatment outcome differences between STAR*D and pivotal clinical trials of investigational drugs.
Despite these limitations, the study also has several strengths. These include a large sample recruited from multiple geographically diverse sites in both primary and psychiatric specialty care settings. Also, measurement-based care
(4,
28) with protocol-driven treatment and systematic collection of data on outcomes and adverse events was used in both samples as a method of standardizing the treatment delivery and outcomes assessment. This procedure mimics rather closely the treatment procedure used in efficacy trials.
In summary, we found numerous baseline differences between the efficacy and nonefficacy samples. In addition, patients in the efficacy group had better outcomes even after adjustment for these differences. Thus, inclusion criteria for phase III trials result in samples that are not fully representative of depressed outpatients typically treated in practice. If phase III trials enrolled more representative patients, the results would provide better estimations of the benefit to be expected in practice. One could also speculate that studying more representative groups might also reduce placebo response rates. However, the less well-documented safety profile of investigational antidepressants would have to be considered in broadening phase III trial inclusion criteria.