The advent of antipsychotic medications acting at dopamine receptors revolutionized the treatment of positive symptoms in schizophrenia. On the basis of the antidopaminergic properties of these drugs, a dopamine hypothesis proposed that the positive symptoms of schizophrenia are due to an excess of dopamine signaling in the striatal and/or mesolimbic areas of the brain (
1). Over time, this theory was modified to also suggest that schizophrenia's negative symptoms are related to deficits in prefrontal cortical dopamine neurotransmission (
2,
3). Hypotheses centered on dysfunction in other neurotransmitter systems—including glutamate, γ-aminobutyric acid (GABA), serotonin, and acetylcholine—also have been proposed (
3,
4). The neurodevelopmental hypothesis posits that schizophrenia's origins lie in the complex interplay of environmental and genetic factors during brain development (
3,
5).
In addition to improved formulations of etiologic theories, continued advances in genomics and molecular biology provide new opportunities to expand our understanding of the cellular/developmental mechanisms of schizophrenia and antipsychotic medications by investigating their effects on neuronal signaling. Here we will focus on two pathways that work in parallel and may intersect—the Akt/glycogen synthase kinase 3 (GSK-3) pathway and the wingless (Wnt) receptor pathway. Work related to these molecules has received considerable attention in the basic scientific literature, but they are relatively new to the psychiatric literature, particularly in relation to clinical applications in schizophrenia. An expanded understanding of how such molecules work will help to advance our ability to identify patients at risk as well as potentially develop new therapeutic approaches. We present the following: 1) an overview of the molecules and related signaling in the Akt/GSK-3 and Wnt pathways, 2) evidence that molecules in the Akt/GSK-3 and Wnt-related pathways are involved in the pathogenesis of schizophrenia, 3) evidence from animal studies demonstrating that alterations in these signaling pathways are functionally significant, 4) the relationships of these pathways to antipsychotic drug action, and 5) predictions and future directions regarding use of these pathways to develop our understanding of schizophrenia and its treatment.
Overview of Signaling Pathways
Akt (known also as protein kinase B) is a protein kinase that phosphorylates substrates on specific serine and threonine residues and is involved in multiple cellular functions including metabolism, cell stress, cell-cycle regulation, and apoptosis (see
Table 1 for definitions of specific terms). Akt has a basic role in regulating neuronal cell size and survival (
6,
7). It modulates synaptic plasticity and thereby may affect brain functions such as long-term potentiation, working memory, and fear conditioning (
7–
10). Akt also plays a role presynaptically, where it mediates intracellular trafficking of biogenic amine transporters including the dopamine and norepinephrine transporters (
11; Aurelio Galli, personal communication). Dopamine-mediated decreases in Akt activity are thought to be mediated by postsynaptic dopamine D
2 receptors (
12), which is the principle focus of this review.
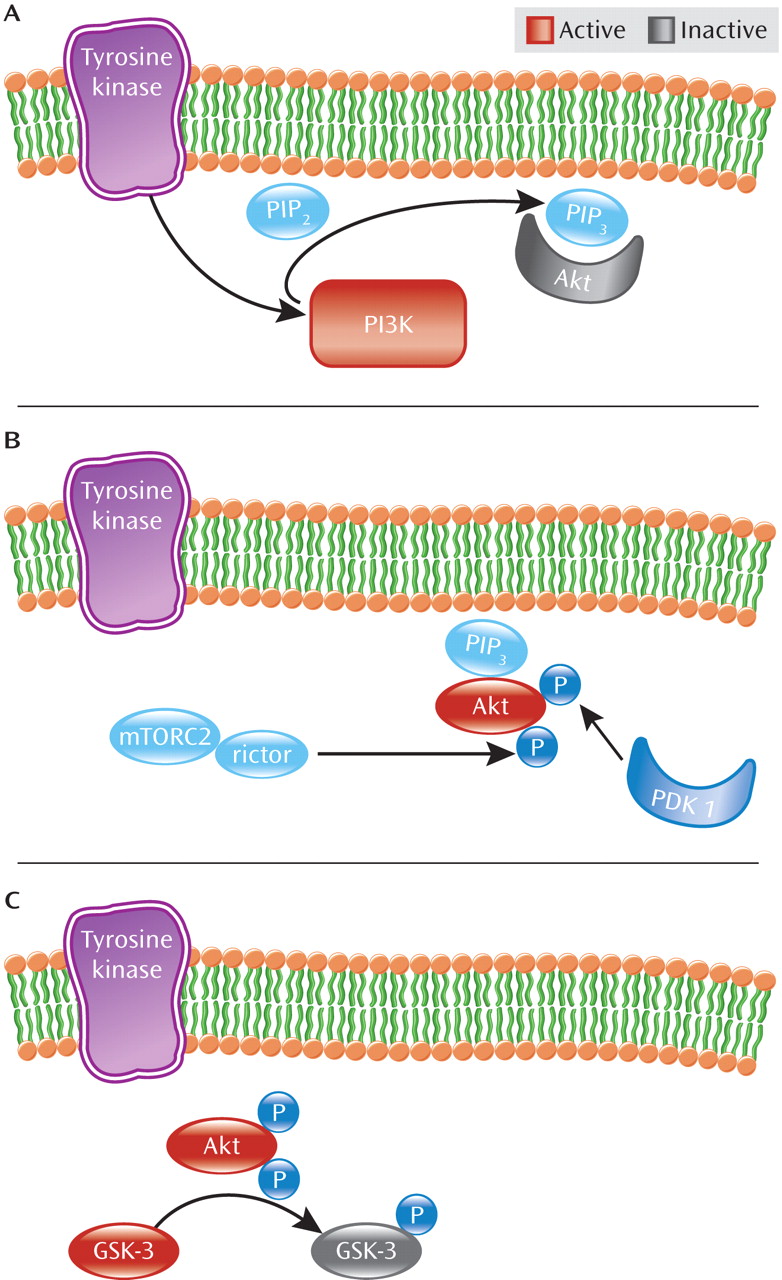
Akt is recruited to the neuronal cell surface by binding to lipids generated by phosphatidylinositol 3-kinase (PI3K). As detailed in
Figure 1, in response to stimulation of plasma membrane receptors including receptor tyrosine kinases, PI3K generates the signaling lipid, phosphatidylinositol 3,4,5 trisphosphate (PIP
3) at the cell surface (
13). This leads to recruitment of Akt to the plasma membrane through its association with the newly generated PIP3. Akt is subsequently activated through phosphorylation by the intracellular kinases PDK1 (3-phosphoinoitide-dependent protein kinase 1) and rictor-mTORC2 (mammalian target of rapamycin complex 2) at positions Thr 308 and Ser 473, respectively (
6,
14). Once active, Akt phosphorylates a number of other molecules including the clinically relevant target, GSK-3—also a protein kinase (
15). Beyond its function in glucose metabolism, GSK-3 plays numerous roles in differentiation and development, intracellular trafficking, apoptosis, and regulation of gene transcription (
16). In the brain, GSK-3, like Akt, may also modulate synaptic plasticity (
17). In contrast to Akt, however, GSK-3 is constitutively active in resting cells, requiring phosphorylation by kinases such as Akt to inactivate it (
18).
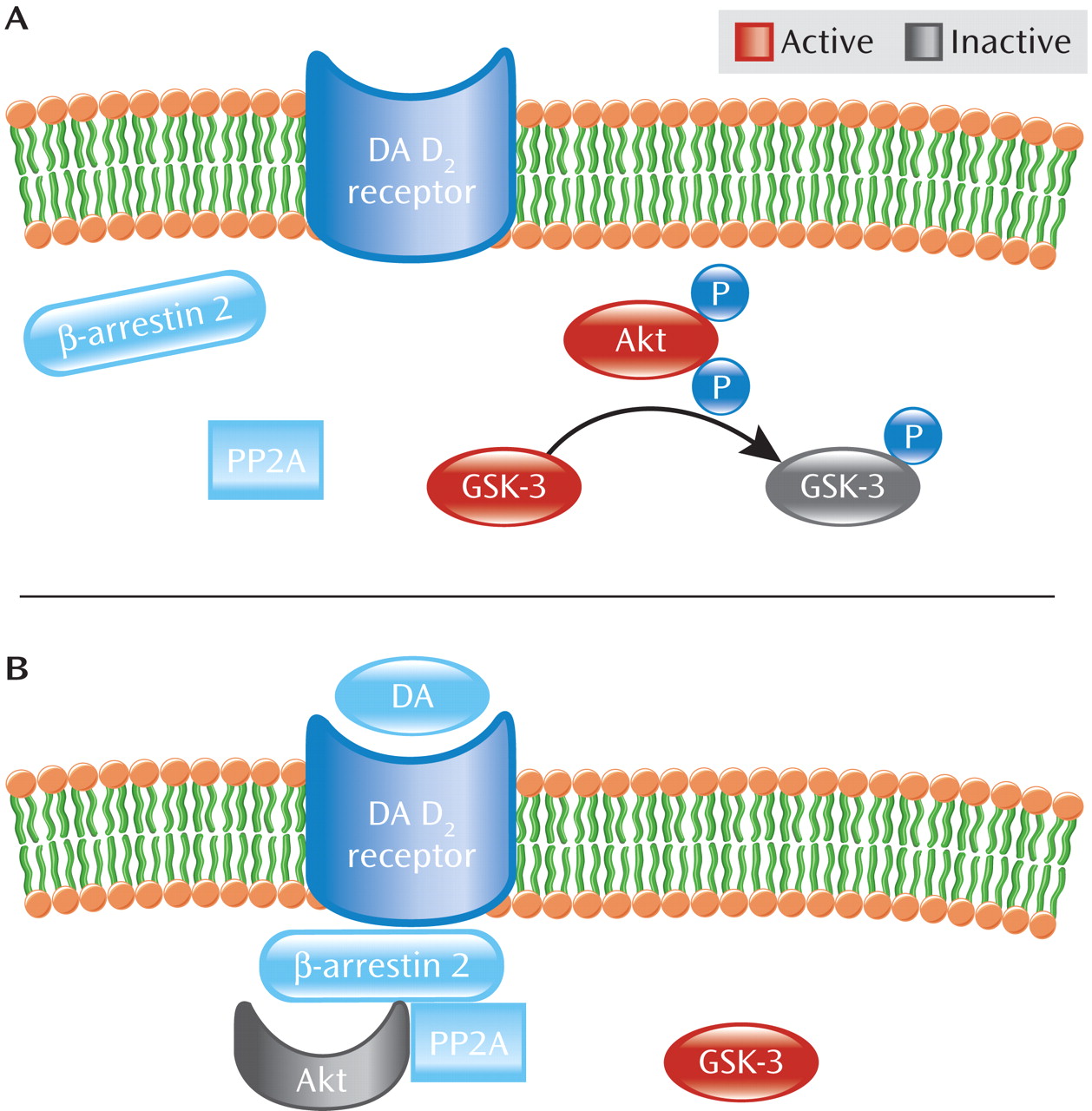
As seen in
Figure 2, Akt activity is further modulated by dopamine D
2 class receptors (D
2–4 receptors) (
12). Activation of dopamine D
2 receptor by dopamine leads to recruitment of β-arrestin 2 (also known as arrestin 3), a scaffolding protein, which facilitates receptor desensitization and subsequent internalization (
19–
21). Akt is also recruited into this complex along with the phosphatase PP2A, which dephosphorylates and consequently inactivates Akt, resulting in increased GSK-3 activity (
19,
22,
23). Thus, dopamine D
2 receptor activation inhibits Akt activity through an arrestin-dependent but G protein-independent pathway (
12,
15,
19). By blocking dopamine D
2 receptor activation, antipsychotic medications prevent β-arrestin 2 recruitment (
20,
24) and enhance Akt activity. Curiously, the mood stabilizer lithium has been shown to disrupt the β-arrestin 2/Akt/PP2A complex, thereby preventing dopamine-induced dephosphorylation of Akt and thus blocking amphetamine-induced locomotion (
25). Moreover, amphetamine-induced locomotion is greatly diminished in β-arrestin 2 knockout mice, suggesting that this pathway is critical to at least some psychostimulant effects (
19).
Three isoforms of Akt have been identified in mammalian cells: Akt1, Akt2, and Akt3 (
26). These isoforms appear to have distinct roles in a variety of processes, including development and metabolism. Mice with only one isoform knocked out are viable, suggesting in vivo compensation by the other two isoforms (
26). Akt1 has been the isoform most associated with schizophrenia and therefore will be the focus of much of the work discussed in this review. Akt2 has been implicated in regulation of metabolism primarily through insulin-regulated glucose homeostasis, while Akt3 plays a role in postnatal brain development (
27,
28). To date, genetic and molecular studies have focused almost exclusively on Akt1 with respect to associations with schizophrenia, leaving roles for Akt2 and Akt3 in the disorder unknown and potentially the subject of future investigations.
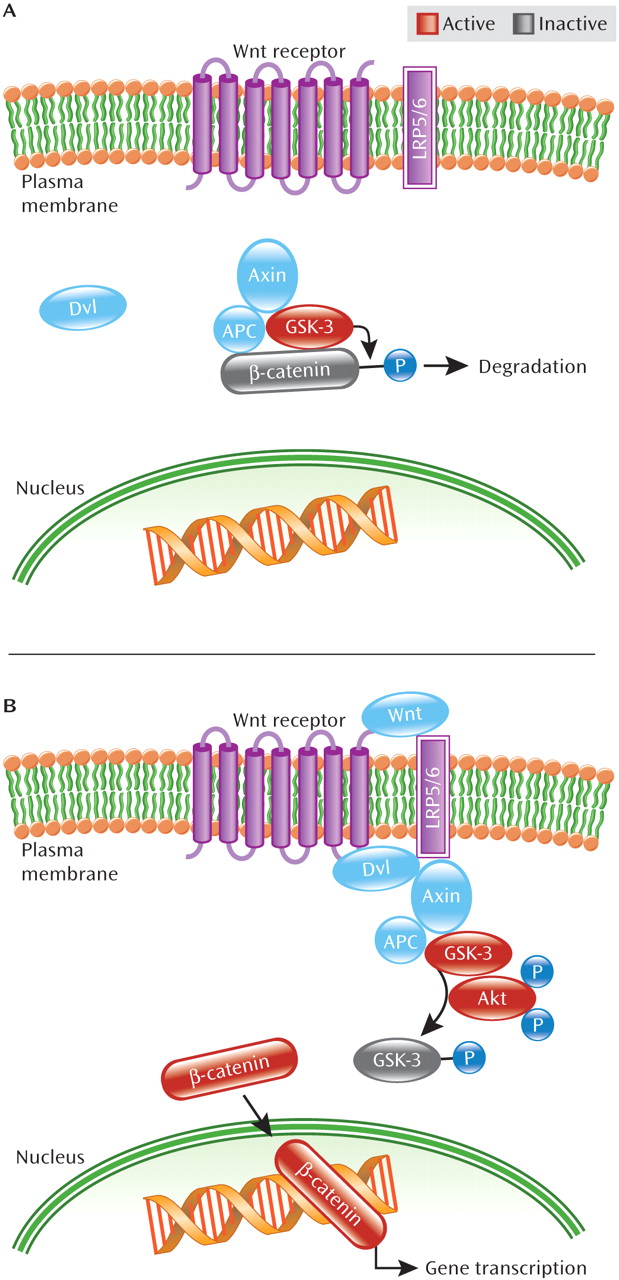
In addition to the Akt/GSK-3 pathway, signaling through the Wnt pathway has been inferred to be of clinical relevance in schizophrenia. Members of the Wnt protein family are secreted glycoproteins implicated in diverse neuronal processes, including brain development, regulation of synaptogenesis and synapse specificity, and oncogenesis and determination of cell fate (
29–
32). Moreover, disruptions in Wnt signaling lead to abnormal brain development in mice (
5,
31). GSK-3 also plays an important role in Wnt signaling. As illustrated in
Figure 3, GSK-3β, the GSK-3 isoform most studied in schizophrenia, exists in a complex with multiple proteins: β-catenin (a transcriptional mediator and cytoskeletal protein), APC (adenomatous polyposis coli, a tumor suppressor), and Axin (a scaffolding protein) (
23). In the absence of Wnt receptor activation, GSK-3β constitutively phosphorylates β-catenin resulting in β-catenin's degradation, thus blocking β-catenin-mediated gene transcription. However, binding of ligands to the Wnt receptor leads to phosphorylation of the Wnt receptor's binding partner—one of two low-density lipoprotein receptor-related proteins, LRP5 or LRP6, as well as activation of the intracellular protein dishevelled (Dvl), facilitating physical separation of GSK-3β from β-catenin (
23). This separation prevents β-catenin's phosphorylation by GSK-3β and allows β-catenin to escape degradation, resulting in its accumulation in the nucleus. Once in the nucleus, β-catenin regulates transcription through interactions with DNA transcription factors, ultimately leading to expression of specific proteins in the cell (
16,
23).
Evidence also points to convergence between the Wnt and Akt/GSK-3β pathways. Activation of the Wnt pathway stimulates Akt to act in concert with Dvl on GSK-3β. It has been suggested that Dvl's interaction with Axin in the Axin/APC/GSK-3β complex leads to increased susceptibility of GSK-3β to be phosphorylated, and thus inactivated, by Akt (
33).
Signaling Pathways and Schizophrenia Pathogenesis
Genetic linkage analyses show cosegregation of Akt1 haplotypes with schizophrenia, suggesting that the
AKT1 gene may be a schizophrenia susceptibility gene (
34). Similarly, genetic studies demonstrated association of
AKT1 gene polymorphisms with schizophrenia in a Chinese population (
35). These associations also have been replicated in other studies, including the Irish Study of High Density Schizophrenia Families (
36). There is also evidence suggesting significant alterations in Akt1 levels in people with schizophrenia. Lymphocytes from individuals with schizophrenia expressed 68% less Akt1 relative to comparison subjects (
34). Importantly, Akt1 was reduced in the hippocampus and frontal cortex in postmortem brain samples from those with schizophrenia relative to comparison subjects; in contrast, Akt2 and Akt3 levels were unaffected (
34). However, differences in Akt1 levels derived from postmortem brain samples must be approached with some caution, given potential variations in levels of intact protein remaining in tissue (dependent in part on the postmortem interval). Additionally, a potential confounding factor in using postmortem brain tissue to ascertain Akt1 levels is whether the lower levels seen in the brains of subjects with schizophrenia is due to the illness or is a consequence of often chronic treatment with antipsychotic drugs. Emamian et al. (
34) attempted to control for effects of chronic drug treatment by using a mouse model of chronic antipsychotic treatment with haloperidol. Future studies comparing Akt1 levels using brain tissue obtained from drug-naive subjects or subjects with other disorders also treated chronically with antipsychotic medications (i.e., bipolar disorder) may help to further clarify the association of brain Akt1 levels with schizophrenia.
Akt may also play a role in cognition through its modulation of synaptic plasticity. Studies have demonstrated Akt's involvement in long-term potentiation and fear conditioning (
8–
10). Akt1 deficiency in mice also results in abnormalities in working memory and changes in prefrontal cortex neuron architecture (
7). Similarly, GSK-3β has been implicated in cognition through its mediation of long-term potentiation and long-term depression (
17). Consistent with these studies, the potential association of genetic variation in the
AKT1 gene with neurocognition in schizophrenia was examined using a large pool of subjects with schizophrenia (N=641) drawn from the multicenter Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) study. Using the five schizophrenia-associated single nucleotide polymorphisms (SNPs) in the
AKT1 gene originally proposed by Emamian et al. (
34), no significant association with multiple neurocognitive domains was found (
37). More recently, Tan et al. (
38) examined the relationship of these five
AKT1 SNPs to cognition in healthy subjects and found that variants of the gene are associated with differences in specific domains of cognitive function including IQ, executive functioning, and processing speed. Furthermore, one of the SNPs was associated with reduced expression of Akt1 in peripheral lymphocytes and brain gray matter. This SNP was also associated with brain volume reductions in the caudate bilaterally and right prefrontal cortex (
38). This is consistent with increasing evidence for Akt-mediated regulation of neuronal cell size and Akt/PI3K-mediated cell volume (
6,
39). In focusing on only five SNPs, however, both of the above studies may have missed other SNPs that do indeed play a role in modulation of neurocognition in schizophrenia
Disruption of Akt's regulation of GSK-3 activity in the brain may also play a role in dysregulation of brain function in disorders such as schizophrenia, and its restoration by antipsychotic medications may contribute to the clinical efficacy of these drugs. Consistent with the finding that Akt1 levels were reduced in the frontal cortex of subjects with schizophrenia, levels of phosphorylated GSK-3β were also reduced in the frontal cortex of people with schizophrenia (
34). However, the relationship between GSK-3 activity and total amounts of GSK-3 in specific brain regions remains unclear. Despite studies demonstrating reductions in overall levels of GSK-3β in the frontal cortex of subjects with schizophrenia, no correlation was found between levels of GSK-3β activity and its levels in the frontal cortices of these subjects (
40,
41). Like Akt1, postmortem brain tissue was used to determine GSK-3 levels and activity, leaving open the potential confound of variations in GSK-3 activity due to differences in the postmortem interval or tissue handling. Alternatively, since only total GSK-3 activity was measured, another GSK-3 isoform, GSK-3α, might have compensated for reduced GSK-3β levels in the frontal cortex. Although the physiological consequences of changing GSK-3β levels and/or activity remain to be determined, it has been proposed that the reduction in GSK-3β levels may affect other signaling pathways such as the Wnt pathway (see below).
If these reductions occur early in life, it may lend further credence to the neurodevelopmental model of schizophrenia (
41). Consistent with this, recent work has linked GSK-3 to Disrupted in Schizophrenia 1 (DISC1), a protein involved in brain development and whose gene was found to be disrupted by a balanced chromosomal translocation in a Scottish family with high incidence of schizophrenia (
42). DISC1 directly interacts with GSK-3, resulting in inhibition of GSK-3β activity and subsequent stabilization of β-catenin. Functionally, DISC1's inhibition of GSK-3β was also shown to regulate proliferation of neuronal precursor cells in both mouse embryonic and adult brains, further arguing for a role for GSK-3 and Wnt pathway signaling in neurodevelopment (
42). However, to date this relationship between DISC1 and GSK-3β has only been demonstrated in mice; similar studies will be needed in humans to make more direct links between DISC1, GSK-3β, and schizophrenia.
As for Akt and GSK-3, Wnt signaling abnormalities have also been demonstrated in schizophrenia. Levels of β-catenin are decreased in the brains of people with schizophrenia, presumably due to aberrant regulation of β-catenin degradation mediated by activated GSK-3β (
5,
23). This is consistent with Emamian et al.'s findings demonstrating enhanced GSK-3β activity secondary to diminished phosphorylation by Akt, since levels of Akt are reduced in regions of the brain in those with schizophrenia (
34). Moreover, increased expression of Wnt-1, an important molecule in the Wnt pathway, has been found in the hippocampus of subjects with schizophrenia relative to comparison subjects (
43). Genetic association analyses have also demonstrated a strong association between the locus for a gene encoding a Wnt receptor, FZD3, and schizophrenia in a Chinese population (
44,
45). Additionally, recent work found associations between genes in the Wnt signaling pathway and psychotic subtypes of bipolar disorder (
46,
47). This further suggests a potential role for the Wnt pathway in psychosis and as a potential target for antipsychotic drugs.
Functional Significance of Signaling Pathway Alterations
The putative roles of Akt/GSK-3 and Wnt pathways in dopaminergic signaling and schizophrenia have been studied in a number of rodent models. In the case of Akt1, consistent with a recent study demonstrating that specific
AKT1 SNPs may impact on brain volume (
38), Akt signaling appears to decrease the size of dopamine neurons in the ventral tegmental area of rats chronically treated with morphine (
48). Behaviorally, Akt1-deficient mice treated with a psychotomimetic agent such as amphetamine exhibited notable reductions in prepulse inhibition during tests of sensorimotor gating similar to the abnormal sensorimotor gating in individuals with schizophrenia (
34). Akt1-deficient mice were also tested for related abnormalities in spatial working memory, given the working memory deficits associated with schizophrenia (
49,
50). Following pharmacological stimulation with the dopamine D2 receptor agonist quinpirole, these mice performed worse relative to mice treated with saline or a dopamine D
1 receptor agonist (
49). Similar abnormalities were also observed in mice lacking Dvl1, an isoform of Dvl; these mice exhibited significant behavioral abnormalities in social interactions including nesting with and grooming other mice as well as sensorimotor gating abnormalities analogous to those in schizophrenia (
51–
53).
Like Akt, GSK-3 activity has also been shown to play an important role in modulating the dopaminergic response to amphetamine. Amphetamine-induced dopamine transporter (DAT)-mediated dopamine efflux and subsequent postsynaptic D
2 receptor stimulation likely results in Akt inactivation and increased GSK-3 activity (
22). An improved understanding of a dopamine-mediated response to a psychotomimetic agent such as amphetamine in animals may also lead to better animal models of dopamine-mediated psychosis. Rats treated with a specific GSK-3 inhibitor, AR-A014418, failed to display amphetamine-induced hyperactivity (
54). Similarly, heterozygous GSK-3β knockout mice (expressing approximately half of wildtype levels of GSK-3β) displayed significantly reduced levels of locomotor activity following amphetamine treatment (
22). Additionally, treatment of DAT knockout mice with multiple GSK-3 inhibitory drugs inhibited the hyperactive behavior of the nontreated DAT knockout mice (
22). By contrast, transgenic mice with a constitutively active mutant form of GSK-3β displayed hyperactive behaviors similar to mice lacking the dopamine transporter, providing further evidence that GSK-3β activity is linked to dopamine signaling (
20,
55). Moreover, in the absence of inhibition of GSK-3β activity by DISC1 as discussed earlier, mice displayed hyperlocomotion in response to novelty (
42). Importantly, the authors believe that this novelty-based GSK-3β-mediated hyperlocomotion may represent a model for studying positive symptoms in schizophrenia (
42).
Relationship of Signaling Pathways and Antipsychotic Drug Action
There is increasing evidence that Akt and GSK-3β-related intracellular signaling may at least partially modulate the ability of antipsychotic medications to treat symptoms of psychosis. After both acute and chronic treatment of mice with haloperidol, levels of active, phosphorylated Akt1 were increased, resulting in phosphorylation of GSK-3β (
34). These findings suggested that the mechanism of haloperidol's action may be mediated in part by the Akt/GSK-3 system. Similarly in humans, haloperidol treatment may compensate for the decreased levels of endogenous Akt1 in the frontal cortex of people with schizophrenia (
34).
In contrast to these findings, haloperidol treatment of cultured rat cortical neurons led to significant decreases in phosphorylated Akt (
56). Similarly, in vitro haloperidol treatment of rat preneuronal PC12 cells led to loss of the phosphorylated form of Akt (
57). The discrepant effects of haloperidol on Akt may result from differences in vitro versus in vivo as the response in intact brain appears to differ from that observed in neuronal or related cell types in culture. In addition, cells in culture are exposed to different concentrations of drug compared to those in intact brain, possibly leading to concentration-dependent differences in signaling pathways affected (
58). There is precedent for these discrepant findings in other cell culture-based work demonstrating that D
2 receptor activation using agonists such as quinpirole or bromocriptine may also lead to Akt phosphorylation, suggesting that the precise mechanisms of dopamine-mediated Akt phosphorylation are complex and remain to be fully elucidated (
59,
60).
Similar to haloperidol, atypical antipsychotics also impact Akt and GSK-3β activities. Treatment with the atypical antipsychotic clozapine results in increased levels of phosphorylated GSK-3β and total β-catenin (including β-catenin's translocation into the nucleus) (
61,
62). However, differences between haloperidol and atypical antipsychotics have emerged in the kinetics of Akt/GSK-3 phosphorylation, the levels of proteins expressed following drug exposure, and the pathway that is preferentially activated (i.e., Akt versus Wnt pathway signaling). Treatment with either haloperidol or clozapine led to phosphorylation of GSK-3α and GSK-3β in rat frontal cortex. However, whereas levels of phosphorylated Akt1 returned to baseline within 1 hour following acute haloperidol exposure, Akt remained phosphorylated after a similar acute clozapine treatment (
63). The prolonged duration of clozapine's effects on Akt relative to a typical antipsychotic such as haloperidol may account for its greater effect on downstream molecules in the Wnt pathway such as Dvl.
Dvl promotes β-catenin signaling in the Wnt pathway by stimulating GSK-3β phosphorylation in the Axin complex (as discussed above) and also facilitates Akt phosphorylation (
33). Given its roles in both Akt and Wnt pathways, signaling through Dvl may be an important property of clozapine's therapeutic efficacy. Indeed, clozapine increased levels of both total and phosphorylated Dvl (
63), as well of the Dvl isoform Dvl-3, which may also be either directly or indirectly physically associated with the dopamine D
2 receptor (
62). Interestingly, overexpression of Dvl-3 in vitro led to changes in levels of phosphorylated GSK-3 and β-catenin analogous to those observed after antipsychotic treatment, providing an additional functional link between dopamine signaling and the Wnt pathway (
62). Future studies examining Dvl both in vivo and in vitro in response to other antipsychotics could determine more definitively whether effects on Dvl are unique to clozapine or a common property of antipsychotics.
Antipsychotics may also exert their effects in a developmentally dependent manner. While adult rats (14+ weeks old) treated with clozapine, risperidone, or haloperidol had increased β-catenin and GSK-3 levels in the ventral midbrain, no changes were observed in adolescent rats receiving the same drug treatment (8–9 weeks old) (
64).
Predictions and Future Directions
The apparent convergence of D
2 receptors and the Wnt receptors onto the Akt and GSK-3 downstream signaling pathways leads us to propose a model in which constituent molecules of the dopamine D
2 receptor pathway may directly and/or indirectly modulate Wnt pathway signaling. This may allow for both receptors to modulate reciprocally the function of either signaling pathway and allows for potential integration of different cellular signals. However, to date, the relationship between these pathways in neurons remains complex, and the relationship we propose is hypothetical. Recent evidence suggests that signaling proteins may have multiple and at times opposite roles dependent on spatial and temporal regulation. For example, while plasma membrane-associated Akt1 facilitates β-catenin-mediated gene transcription by promoting translocation of β-catenin into the nucleus, nuclear Akt1 inhibits β-catenin-mediated transcriptional activation, demonstrating that much work remains in elucidating relationships not only between signaling pathways but within them as well (
65). Based on the evidence discussed in this article, potential interactions (i.e., direct versus indirect associations) between D
2 and Wnt receptors and possible points of intersection within these pathways will require clarification using a combination of genetic, imaging, and biochemical studies. We also propose that differences in the clinical efficacy of individual antipsychotic medications may result from their respective abilities to modulate Akt activation in the D
2 receptor pathway and/or molecules in the Wnt pathway. Consistent with this, although acting on multiple receptor systems, clozapine's superior clinical efficacy may stem from its ability to act downstream of these receptors on molecules in both Akt/GSK-3 and Wnt pathways and thereby modulate from both pathways (
62). Future studies can examine whether the efficacy of particular drugs is related to effects on levels and activities of proteins in both Akt/GSK-3 and Wnt pathways as opposed to only within one pathway in vitro and in vivo using tissue culture and animal models.
Since the advent of antipsychotic medications, there have been many speculations about the precise mechanisms responsible for the clinical effects of these drugs. While the dopamine hypothesis provided an early framework for contextualizing the mechanisms of antipsychotics, blocking dopamine receptors using these medications is only the beginning of a chain of signaling into which we are just now beginning to gain insight. It is not yet clear which signaling events downstream of the receptors are critical for the therapeutic actions of antipsychotics and which lead to side effects (i.e., metabolic syndrome). However, although the mechanisms and targets for these phenomena are unknown, these drug effects must result from modulation of intracellular signaling pathways. Based on the work presented, signaling through Akt/GSK-3 and Wnt pathways appears to be a common point of intersection for many of the antipsychotics tested, both among typical and atypical classes of medication. Moreover, recent work has also implicated the Akt/GSK-3 pathway in the mechanism of action for the mood stabilizer lithium, suggesting that lithium-induced inhibition of GSK-3 may be critical for modulating its cellular effects (
22,
25). Nevertheless, despite the progress made in understanding the signaling pathways described above, much work remains in determining the extent to which these pathways are involved in schizophrenia. Future studies will need to apply the findings derived thus far from the in vitro studies and animal models described in this review to clinical studies of schizophrenia. Applying increasingly sensitive and sophisticated genome-wide screening techniques may yield new gene polymorphisms associated with schizophrenia that would facilitate verifying or refuting roles for Akt1, GSK-3β, and proteins in the Wnt pathway. Taken together, the increasing understanding of the Akt/GSK-3 and Wnt signaling pathways may create new avenues in development of novel drug therapies. While it is likely that there are multiple pathways implicated in the pathogenesis and treatment of psychosis, the Akt/GSK-3 and Wnt pathways appear to be promising and interconnected mechanisms that may further elucidate the complexities of schizophrenia.
Dr. Freyberg reports no financial relationships with commercial interests. Dr. Ferrando reports receiving speaker's honoraria from Pfizer and AstraZeneca Pharmaceuticals. Dr. Javitch reports consulting fees from Heptares Therapeutics.
This work was supported in part by NIH grants DA-022413 and MH-18870 and the Lieber Center for Schizophrenia Research and Treatment.