Deficits in cortical γ-aminobutyric acid (GABA) neurotransmission are among the most consistent findings in schizophrenia research. These findings support the hypothesis that dysregulation of inhibitory interneurons and/or GABA neurotransmission may contribute to the pathology of this disorder. Postmortem studies demonstrate reduced expression of the 67-kDa isoform of glutamic acid decarboxylase (GAD67), the primary enzyme in GABA synthesis. While interneurons represent approximately 20% of cortical neurons, they are a heterogeneous population that varies in morphology, electrophysiological properties, laminar distribution, innervation of pyramidal neurons, and expression of biochemical markers (
1,
2). In schizophrenia, GAD67 mRNA is down-regulated in only 25%-35% of GABA-ergic neurons in layers II-V of the dorsolateral prefrontal cortex. GAD67 expression in other GABA-ergic cells is maintained at normal levels (
3,
4), which suggests that a subset of interneurons may be affected more severely. Another often replicated finding in schizophrenia is a reduction in parvalbumin (
5), particularly in layers III and IV of the dorsolateral prefrontal cortex. In addition, a reduction in GABA transporter-1 and parvalbumin in cartridges on the axon initial segment of pyramidal neurons has implicated the chandelier subclass of interneurons in schizophrenia (
6,
7). The GAD67 mRNA deficit is also present in layers II and V (
8), however, which suggests that additional interneuron subtypes may also be involved in schizophrenia. Recently a deficit in somatostatin mRNA was identified in schizophrenia, particularly in layers II and III (
9–11), and a reduction of neuropeptide Y mRNA in the dorsolateral prefrontal cortex (
9) has implicated Martinotti and bitufted cells. In addition, cholecystokinin mRNA was reported to be deficient in schizophrenia (
9,
12). These alterations may represent a failure to reach normal expression levels during development or a failure to maintain normal levels of expression during adulthood.
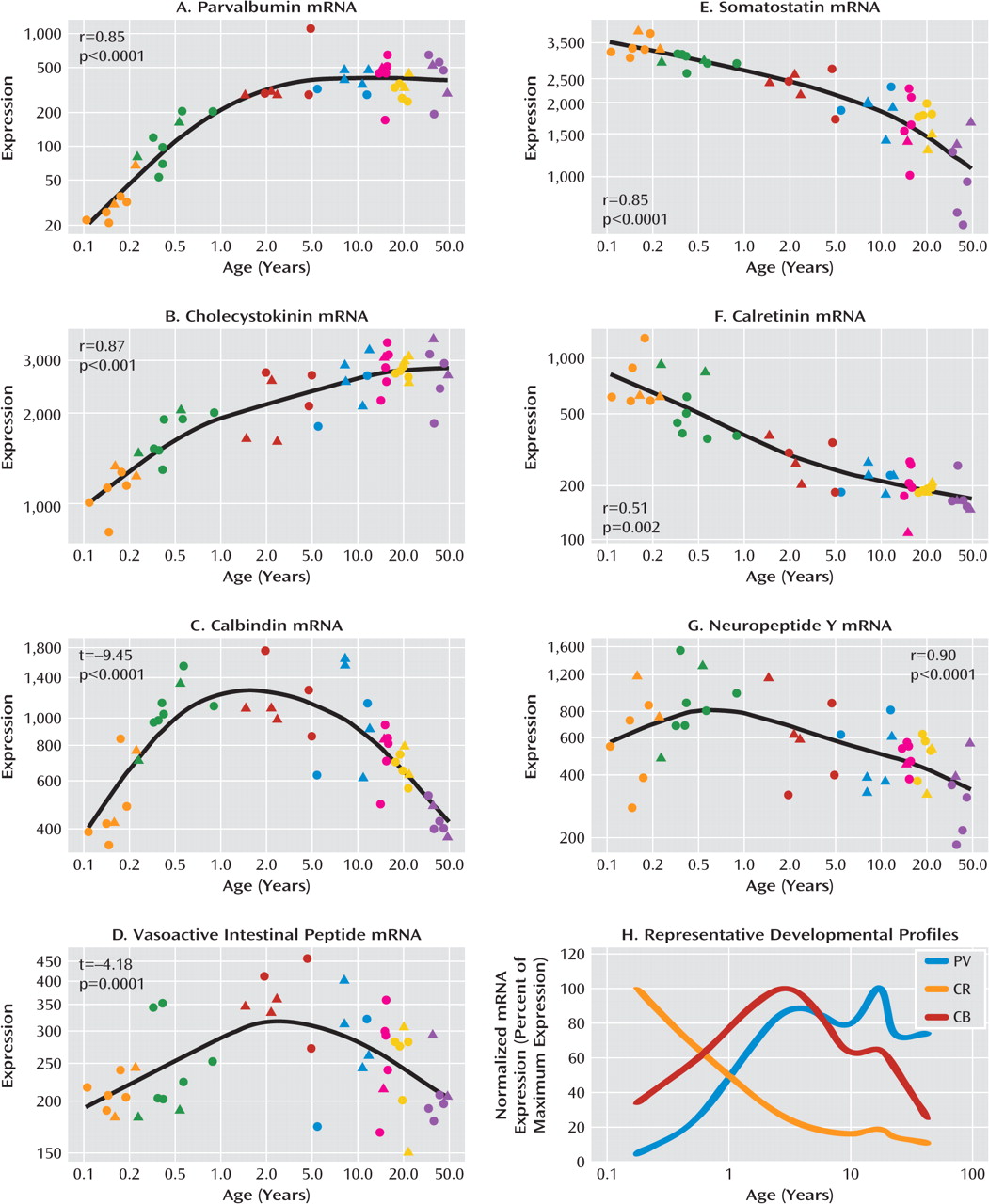
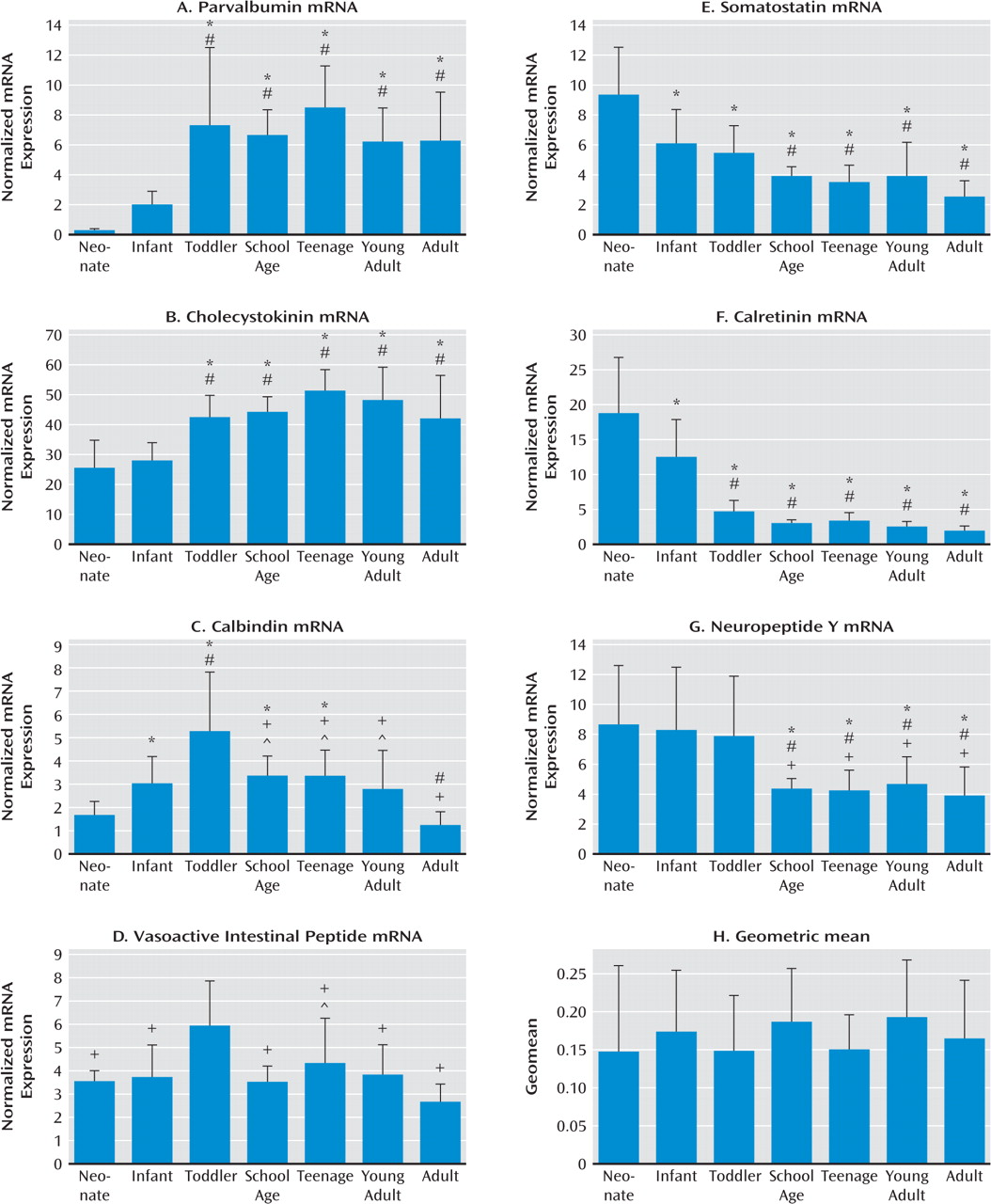
We measured the mRNA expression of seven calcium-binding proteins and neuropeptides expressed by GABA-ergic interneurons (parvalbumin, cholecystokinin, calbindin, vasoactive intestinal peptide, somatostatin, calretinin, and neuropeptide Y) in postmortem dorso-lateral prefrontal cortex tissue of individuals from age 6 weeks to 49 years as well as in an additional cohort of comparison subjects and patients with schizophrenia. We found that interneuron marker genes followed one of three general expression profiles: either increasing or decreasing in expression over postnatal life, with the most dramatic changes in the first few years before reaching a plateau; or a dynamic pattern increasing to peak expression in the toddler years. The development of interneuron markers is thus protracted in the human dorsolateral pre-frontal cortex, extending well into the toddler years and, for some markers, changing during the transition from the teen years to adulthood. Alterations in all seven markers in schizophrenia implicate multiple GABA-ergic interneuron subtypes in the pathology, and although altered markers do not necessarily share the same temporal pattern of maturation or the same spatial origin in development, interneurons implicated in schizophrenia may constitute a subgroup that is dependent on similar upstream growth factors for differentiation and/or survival.
Discussion
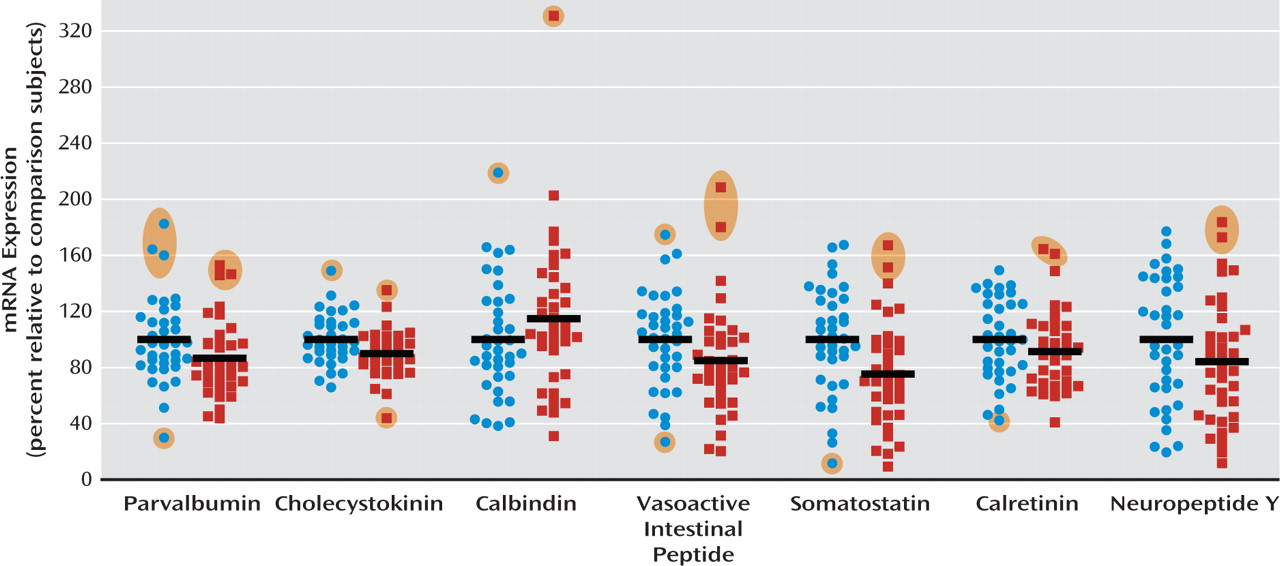
A cortical GABA-ergic deficit is one of the most consistently reported findings in people with schizophrenia. We demonstrate here that six of seven biochemical marker mRNAs for GABA-ergic interneurons were reduced by 10%– 30% in the dorsolateral prefrontal cortex of patients with schizophrenia, with the most marked reduction observed in somatostatin mRNA. We also found that the development of interneuron markers is protracted in the human dorsolateral prefrontal cortex, extending well into the toddler years and, for some markers, extending beyond the teenage years. We considered three possible explanations for the alteration in interneuron markers in schizophrenia: that the interneurons that were most affected 1) share a common developmental origin and may be derived from the same aberrant precursor population; 2) demonstrate a common ontological course, whereby interneurons with protracted development would be most altered in the disease; or 3) may be dependent on a common growth factor that regulates their differentiation or survival.
Our first consideration is that altered expression of multiple interneuron marker mRNAs may indicate dys-regulation of a common precursor cell or of cells from a common developmental origin in schizophrenia. The main source of cortical interneurons in the rodent is the ganglionic eminence. It is thought that the majority of cortical interneurons arise from the medial and caudal ganglionic eminences. Parvalbumin (in axon and soma targeting chandelier and basket cells), somatostatin (primarily expressed in dendrite-targeting Martinotti cells), and calbindin (expressed in multiple subtypes) positive inter-neurons are produced in the medial ganglionic eminence (
16). Neuropeptide Y and calretinin (expressed in several interneuron subtypes) interneurons primarily originate from the dorsal medial ganglionic eminence (
17,
18), and vasoactive intestinal peptide (dendrite-targeting double bouquet and bipolar interneurons) and some calretinin positive neurons are derived from the caudal ganglionic eminence (
19). In the primate cortex it has been suggested that interneurons, particularly calretinin-expressing inter-neurons, may also originate from the dorsal neuroepithelium of the lateral ventricle (
20–22). Thus, the interneurons that are most altered in schizophrenia originate primarily from the medial ganglionic eminence, with some contribution from the caudal ganglionic eminence.
Given the developmental nature of schizophrenia, with the prodrome and disease-related symptoms often manifesting in adolescence or early adulthood (
23), we next considered that late-maturing interneuron subtypes may be particularly vulnerable in schizophrenia. Our findings indicate that interneuron subtypes continue to differentiate and mature through the second decade of life and demonstrate peak mRNA expression of parvalbumin and cholecystokinin in the teenage years, consistent with previous reports (
24,
25). Calbindin and vasoactive intestinal peptide mRNA show similar up-regulation during early development, with a peak in expression during the toddler years, and then decline from the teenage to the adulthood years. Indeed, mRNA expression of cholecystokinin, parv-albumin, and vasoactive intestinal peptide were reduced in schizophrenia, in accordance with reports of these reductions in the frontal cortex of people with schizophrenia (
8–10,
12). Our finding of increased calbindin mRNA expression, however, is confounded by the expression of calbindin by pyramidal cells in layer III (
26,
27) and is not widely replicated in schizophrenia; some studies have reported no change in calbindin immunopositive cell density (
28) and others have reported reduced calbindin-positive cell density in layer II of the prefrontal cortex (
25,
29,
30). The homogenate-based approach used here cannot distinguish whether an increase in calbindin expression may be attributed to interneurons and/or pyramidal neurons; in situ hybridization is required to determine this. One report, however, does support an increased density of calbindin-positive local circuit interneurons across all cortical layers, by 53% and 69% in Brodmann's areas 9 and 46, respectively, attributed particularly to changes in layers III and V/VI in the prefrontal cortex (
31).
Consistent with our hypothesis, late-maturing interneuron markers displaying a delayed increase (parvalbumin and cholecystokinin) are altered in schizophrenia, as found in this study as well as others (
9–11). Additionally, we found that interneuron marker mRNAs that decrease across post-natal life are reduced in schizophrenia, the most dramatic being somatostatin. Consistent with reports in developing primate frontal cortex, where high levels of somatostatin occur in the embryonic brain and decline to adult levels by postnatal day 60 (
32), we found that somatostatin mRNA expression was gradually down-regulated over human postnatal development, although this reduction appears to occur over years. When outliers were excluded in our study, we observed a modest reduction in calretinin mRNA in schizophrenia, whereas prior studies have reported no alteration in calretinin mRNA in the dorsolateral pre-frontal cortex by polymerase chain reaction (
10) or by in situ hybridization (
8). This discrepancy may reflect the increased statistical power in our cohort (37 patients and 37 comparison subjects), which is more than double the size of other cohorts used in calretinin mRNA studies (12 pairs [8] or 15 pairs [10] of patients and comparison subjects) or may be due to differences in the cohorts studied. We did not observe a change in calretinin protein, which is consistent with other studies that found no change in the density of calretinin immunopositive cells (
25,
28,
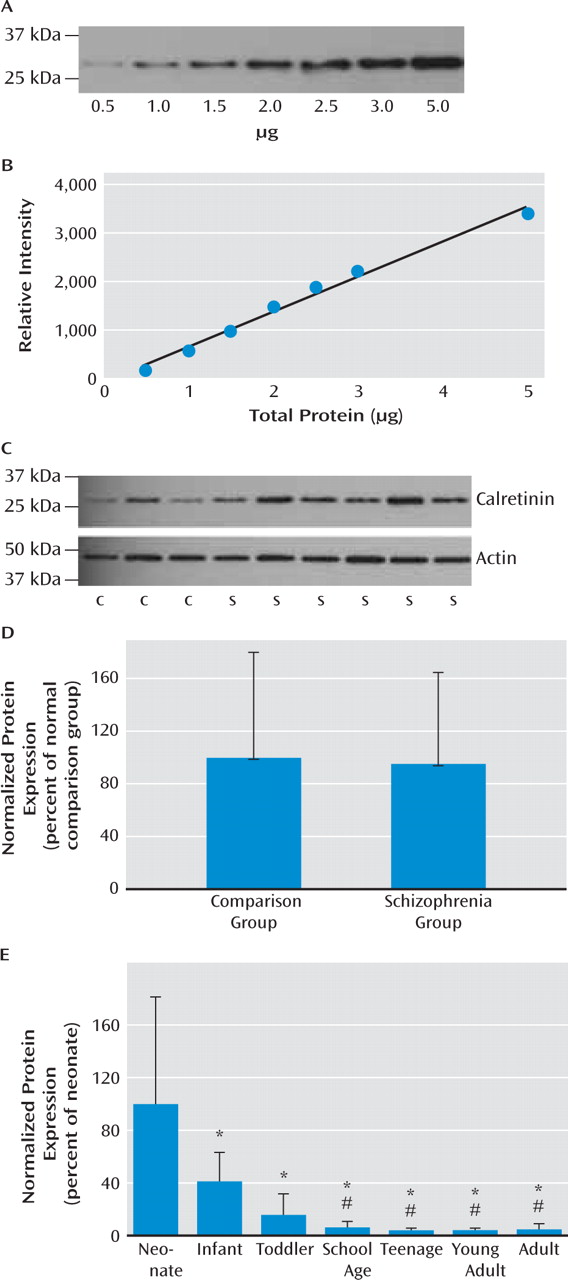
29). Significantly reduced expression of somatostatin (and of neuropep-tide Y and calretinin with the outliers excluded) mRNAs in schizophrenia suggest that not only markers indicative of late maturing interneuron subtypes but also interneuron mRNAs that are abundant during the early phases of life are reduced in schizophrenia. These results suggest that the interneuron deficit may be widespread and could indicate that the underlying mechanisms may be varied, such as a lack of early developmental induction in some individuals and/or a premature age-related decline in others.
Congruent with our third consideration, that aberrant expression of interneuron marker mRNAs may be triggered by alterations in a common upstream pathway, is the finding that expression of brain-derived growth factor (BDNF) and its receptor (TrkB), which are key in the differentiation and survival of interneurons, are reported to be altered in schizophrenia (
33–35). BDNF and TrkB have been shown to up-regulate neuropeptide Y, somatostatin, and cholecystokinin expression in cortical neurons in vitro and in vivo (
36–40). We observed reductions in somatostatin, neuropeptide Y, and cholecystokinin, which would be consistent with an impairment in BDNF signaling. Additionally, mice hypomorphic for TrkB demonstrate a reduction in GAD67 and parvalbumin expression (
33) and have fewer somatostatin-positive cells (
11), which suggests that BDNF-TrkB signaling may represent a common pathway for the differentiation of multiple interneuron subtypes, providing a feasible link to deficient interneuron differentiation or maintenance in the disease state, as has been suggested previously (
33). Our results implicating multiple interneuron subtypes in the cortical pathology of schizophrenia suggest that inhibitory regulation of pyramidal cells may be affected at multiple subcellular levels. For example, parvalbumin-expressing chandelier cells target the axon of pyramidal cells, also implicated by reductions in GABA transporter-1 cartridges on axons and increased GABA
A receptor α
2 subunit expression in the axon initial segment (
5). In contrast, somatostatin, calretinin, and neuropeptide Y positive cells generally target the dendrites of pyramidal cells. Our recent finding (
41) showing a significant reduction in the mRNA encoding GABA
A receptor α
5 subunits, enriched in the dendrites, suggests that GABA-ergic abnormalities may occur pre- and postsynaptically on the dendrites of glutamatergic neurons. The reduction of six interneuron markers and an increase in calbindin mRNA levels in schizophrenia suggest that no one interneuron subtype or developmental profile appears to be particularly vulnerable in schizophrenia, although it appears that calretinin interneurons, but not necessarily vasoactive intestinal peptide interneurons, arising from the caudal ganglionic eminence are the least affected in schizophrenia, as a reduction in calretinin is most inconsistently changed across studies and not confirmed by protein expression in our study. These findings suggest that deficits in GABA-ergic interneurons may be commonly linked through upstream pathways that regulate their differentiation, such as BDNF-TrkB signaling, although further studies are needed to support this hypothesis.