We are presently in transition from genome-wide association studies (GWAS) to "next-generation" sequencing, which will include whole-genome sequencing and other new methods (
1–
3). Although GWAS have failed to explain most of the heritability of human diseases, they have produced a plethora of small but statistically significant associations between genetic polymorphisms and disease. Which of these associations merit pursuit?
Many people would answer, "Results based on large sample sizes and replicated in other large samples." We agree that sample size is very important, but it is not all that matters. GWAS results are vulnerable to bias that cannot be removed by infinitely large samples or even multiple replications using the same method. One such concern is the selection of control subjects.
Unfortunately, selection of control subjects for GWAS is not usually explained in sufficient detail to examine sources of bias. In this respect, the article by Sanders et al. in this issue represents a significant advance (
4). The authors spell out the selection of the control sample for the Molecular Genetics of Schizophrenia Project (MGS2) and compare the prevalences of demographic characteristics, ancestry, and disorders of the control sample with those of the U.S. population. By doing so, they make it possible to scrutinize—and avoid—potential bias in studies that use these controls for GWAS (or other case-control studies). This is a tremendous service to the investigators who will use the MGS2 control sample.
The potential for "population stratification" bias due to subtle differences in the ancestry of case and control subjects is well recognized. It is difficult to entirely remove this bias in GWAS (
3). In psychiatric research, another potential bias is often introduced by following a misguided strategy to remove bias: the use of well (or "hypernormal") control subjects. This strategy is implemented by excluding control (but not case) subjects who have psychiatric disorders other than the one being investigated (
5,
6), and it requires considerable effort and expense. Ironically, as we show here, it is far more likely to cause than to alleviate bias.
In a GWAS, the goal is to identify genetic variants that are causes of (or at least proximate to causes of) the disorder under investigation. Suppose we conduct such a study of schizophrenia using a hypernormal control group. When we analyze the data, we find a genetic variant, A, that has a very small association with the disorder but passes the rigorous significance level of a large GWAS. A similar result for genetic variant A and schizophrenia is subsequently replicated in three further large GWAS of similar design. Is this sufficient to conclude that genetic variant A is a risk factor for schizophrenia?
We propose that it is not. If the result for genetic variant A is valid, it should also be replicable in a more rigorous test: a large longitudinal cohort study with complete follow-up and case ascertainment in which we compare the risk of schizophrenia for individuals with and without genetic variant A. If we were to implement this more rigorous test for genetic variant A, we would replicate the result of a GWAS with an appropriate control group, but we would not necessarily replicate the result of a GWAS that used hypernormal control subjects. To demonstrate this point, we use a hypothetical example.
Example
GWAS With Appropriate Control Group
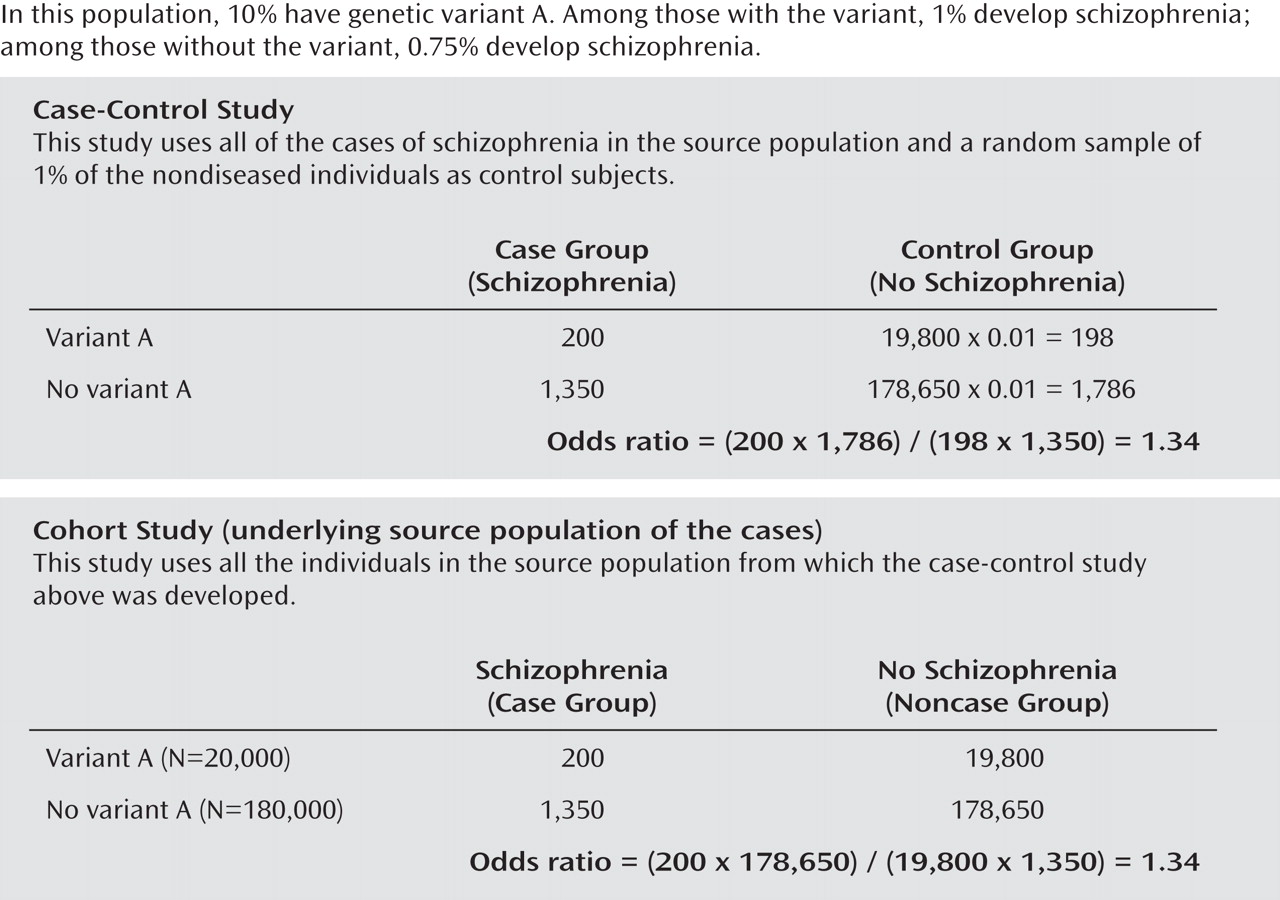
We begin with a study population of 200,000 individuals ages 35 to 54 within a defined and stable community. Ten percent (20,000) have genetic variant A, and 90% (180,000) do not. The proportion with schizophrenia is 1% among individuals with genetic variant A and 0.75% among individuals without genetic variant A. (To focus on the issue of control subject selection we made several simplifying assumptions, including no other sources of bias).
We conduct a GWAS in this study population (
Figure 1, upper part). Our control subjects are a 1% random sample of all subjects without schizophrenia ages 35–54 in this population. We compare the 1,550 case subjects and 1,984 control subjects with respect to 500,000 genetic variants. For genetic variant A, we find an odds ratio of 1.34, which is large for a GWAS, and a p value of 0.007. (If we increased the sample size by a factor of 10, the p value would be less than 0.0000000). As shown in the lower part of Figure 1, if genetic variant A is detected in our GWAS, it will also be detected in our cohort study and the odds ratio will be the same, 1.34.
GWAS With Hypernormal Control Group
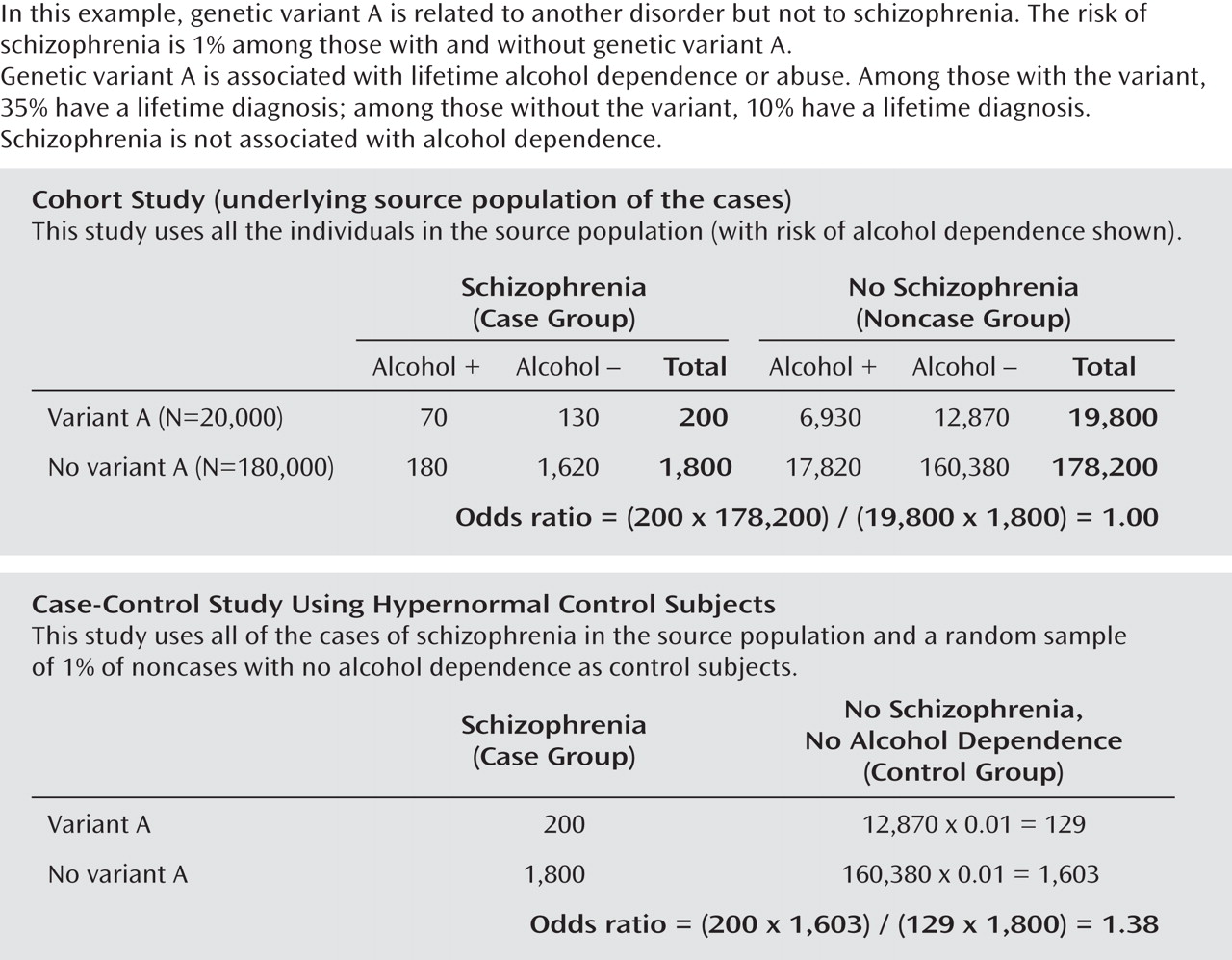
As before, we begin with 200,000 individuals ages 35–54 years, of whom 10% (20,000) have genetic variant A and 90% (180,000) do not. By contrast with the previous example, however, there is
no association between genetic variant A and schizophrenia (the true odds ratio is 1). But there is an association between genetic variant A and alcohol abuse or dependence. The proportion with a history of alcohol abuse or dependence among those with A is 35%, and among those without variant A it is 10%. As shown in the upper part of
Figure 2, a cohort study of the complete source population will yield the correct odds ratio of 1.00 for schizophrenia.
Now we conduct a GWAS in this population. We use all the subjects with schizophrenia and select for control subjects a 1% random sample of the subjects without schizophrenia who do not have a history of alcohol abuse or dependence. As shown in the lower part of Figure 2, we obtain an odds ratio of 1.38 for genetic variant A and schizophrenia, when the true association is null. The odds ratio of 1.38 is of similar magnitude and statistical significance to the odds ratio of 1.34 we found in the previous GWAS with an appropriate control group. But in this instance the statistically significant GWAS result for genetic variant A is entirely an artifact of the process for selecting control subjects. The artifact cannot be removed by increasing sample size or replicating the result in other studies with hypernormal control groups.
What Is a Case-Control Study?
A modern understanding of a case-control study conceptualizes it as an efficient way to sample an underlying cohort of exposed and unexposed people some of whom develop the disease of interest (
7,
8). This is most clearly seen in the context of a nested case-control study in which the underlying cohort is enumerated—but the same logic applies to all case-control studies. The goal of a case-control study is to obtain the result that one would have obtained in a perfect cohort study but with far fewer respondents.
Here we ignore sampling variation for the moment to focus on the principles of validity rather than power.
The validity of the case-control study depends on two critical steps: 1) selecting control subjects from the same source population that gave rise to the cases and 2) selecting control subjects independent of exposure status. When these principles are applied, the control subjects will represent the ratio of exposed to unexposed (those with and without the genetic variant) in the population from which the cases were derived.
The control subjects in the upper part of Figure 1 recreated the ratio of "A" to "not A" among all the noncase subjects in the population. This ratio was 0.11 (198/1,786) in the case-control study and 0.11 (19,800/178,650) in the underlying cohort study. By contrast, the selection of hypernormal control subjects exhibited in the lower part of Figure 2 violated the principles for valid control subjects by imposing restrictions on the control subjects (i.e., exclusion of individuals with alcohol abuse or dependence) that were not imposed on the case subjects. The ratio of "A" to "not A" was 0.08 (129/1,603) in the case-control study and 0.11 (19,800/178,200) in the cohort study. That is how the artifact was introduced. It cannot be removed by increasing sample size or by replication with the same method. More generally, if any genetic variants cause, or are proximate to genetic variants that cause, the disorders excluded from the control but not the case group, the odds ratio will be greater than 1 for these variants. This bias will arise whether or not the disorder being investigated (in our example, schizophrenia) is comorbid with the disorder or disorders excluded from the control group (in our example, alcohol abuse or dependence).
Implications
We do not suggest that GWAS results should be considered invalid whenever hypernormal control subjects were used. When there are no genetic polymorphisms that have a strong effect on the disorders excluded, the bias can be ignored or will at least be minimal. We cannot simply assume, however, that there are no polymorphisms with strong effects on the excluded psychiatric disorders. There is in fact a genetic polymorphism (of ALDH2) that has a strong (inverse) association with alcohol abuse and dependence and is common in Japan and China (
9).
We suggest, therefore, that the results from such studies need extra scrutiny. Sanders et al. (
4) have provided the kind of data required for such scrutiny. When the data are not available, the question can be addressed only by simulations that make various assumptions about the relationships among genetic variants and other disorders.
With regard to the coming era, it is too early to know what the dominant designs will be. It seems likely, however, that with increased precision and detection of rare variants, the magnitude of effects will be larger. This means that the potential bias introduced by using hypernormal control groups will be larger. In this era, investigators could readily avoid the burdensome and ineffective practice of selecting hypernormal control subjects. More generally, we suggest that the selection of appropriate control subjects should receive more systematic attention than it has in the era of GWAS. The precondition for progress toward this end is to articulate for readers the selection of case and control subjects, as exemplified for the control group in the article by Sanders et al. (
4).