Neurodevelopmental disorders such as fragile X syndrome and autism spectrum disorders (hereafter referred to as “autism”) (
1–3) are increasingly linked to synaptic abnormalities (“synaptopathies”). Recent evidence indicates that copy number variants in synaptic genes tend to be overrepresented in neurodevelopmental disorders (
4–6). Postsynaptic function is controlled by a highly complex, interconnected network of molecules (
7). However, a specific synaptic pathway comprising the predominantly presynaptic neurexins (NRXN) and their postsynaptic binding partners, neuroligins (NLGN) and SHANK (
8), has been implicated in autism. Human loss-of-function variants in
NLGN3,
NLGN4,
NRXN1, and
SHANK3 are associated with autism (
3). In mice,
Nlgn3 and
Nlgn4 null mutation have been found in some studies to cause autism-like traits (for example, references
9,
10–12), while
Shank1 deletion has been found to alter dendritic spine size and learning (
13).
PSD-95 (postsynaptic density-95,
DLG4), a member of the membrane-associated guanylate kinase family of synaptic molecules, serves as a major functional bridge interconnecting the neurexin-neuroligin-SHANK pathway (
7) and orchestrates protein-protein interactions and receptor stabilization at excitatory synapses (
14). Gene variation and gene deletion of another family member, SAP102 (
DLG3), is associated with X-linked mental retardation in humans (
15) and autism-like abnormalities in mice (
16), respectively. Microdeletion of chromosomal region 3q29 containing the membrane-associated guanylate kinase SAP-97 (
DLG1) is also associated with autistic traits (
17). However, a possible role for PSD-95 in autism and other neurodevelopmental disorders has not been studied.
Discussion
Our a priori hypothesis was that DLG4 deletion would produce phenotypic abnormalities relevant to neurodevelopmental disorders, particularly autism. We found that DLG4−/− exhibited a set of behavioral, dendritic spine, and molecular abnormalities, some of which were autism-like, but others that are more related to other neurodevelop-mental disorders, notably Williams' syndrome. Furthermore, we found that DLG4 variation was associated with signature neural endophenotypes of Williams' syndrome in a normal human population.
Autism is characterized by repetitive and stereotypic behaviors (
19,
21). On a T-maze test, which measures the tendency of rodents to spontaneously alternate exploration of two goal arms,
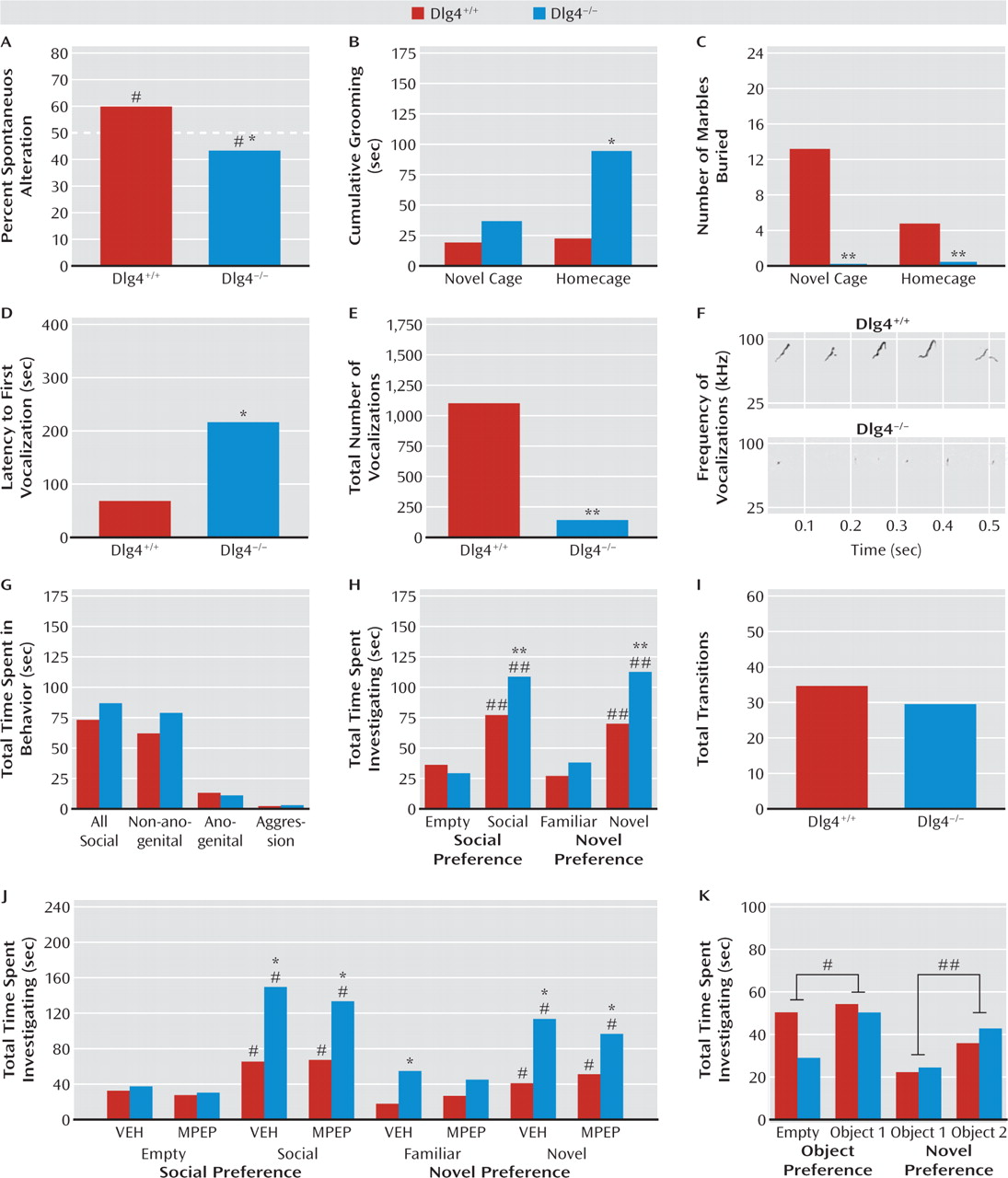
DLG4−/− exhibited modest but statistically significant repetitive exploration of the same arm.
DLG4−/− also displayed excessive grooming behavior (as also found in putative mouse autism models [
27–29]). Notably, this was seen only in a homecage setting and not in a novel context, possibly because of response competition with the suppression of behavior in novel environments we observed in the mutants (see below). This could also possibly account for the decrease, rather than repetitive-like increase, in marble burying in
DLG4−/−, although that was seen in both novel and homecage environments. Nevertheless, these data show that
DLG4 deletion produced some autism-like repetitive, stereotypical behaviors in both the cognitive and noncognitive realms.
Abnormal communicative and social behavior are cardinal features of autism. Male
DLG4−/− vocalized less in the presence of a proestrous female—a phenotype thought to be salient to communication deficits in autism (
19).
DLG4 deletion did not, however, produce an autism-like reduction in social behavior, either during a free dyadic encounter or in a social approach test (
19). Unexpectedly,
DLG4−/− actually spent more time investigating the social and novel-social stimulus than
DLG4+/+. Demonstrating that this increased social investigation was not confounded by
DLG4−/− simply moving less in this test context (as they do in novel, nonsocial contexts—see below), genotypes did not differ in the number of transitions between stimuli. Furthermore, when we replaced social stimuli with inanimate objects in the social approach procedure,
DLG4−/− did not show increased investigation of nonsocial objects, indicating that a general preference for novelty per se did not drive the social phenotype in the mutants. Thus, these data point to an unanticipated increase in certain social behaviors in
DLG4−/−. Although previous studies have shown that treatment with MPEP can prevent seizures and sensorimotor gating deficits in a mutant mouse model of fragile X syndrome (
1), we found that MPEP treatment did not decrease social behavior in
DLG4−/− to
DLG4+/+ levels. Recent work has shown that social abnormalities in a model of autism are similarly insensitive to this treatment (
22), and additional studies are needed to identify pharmacological and behavioral manipulations that effectively normalize such disturbances.
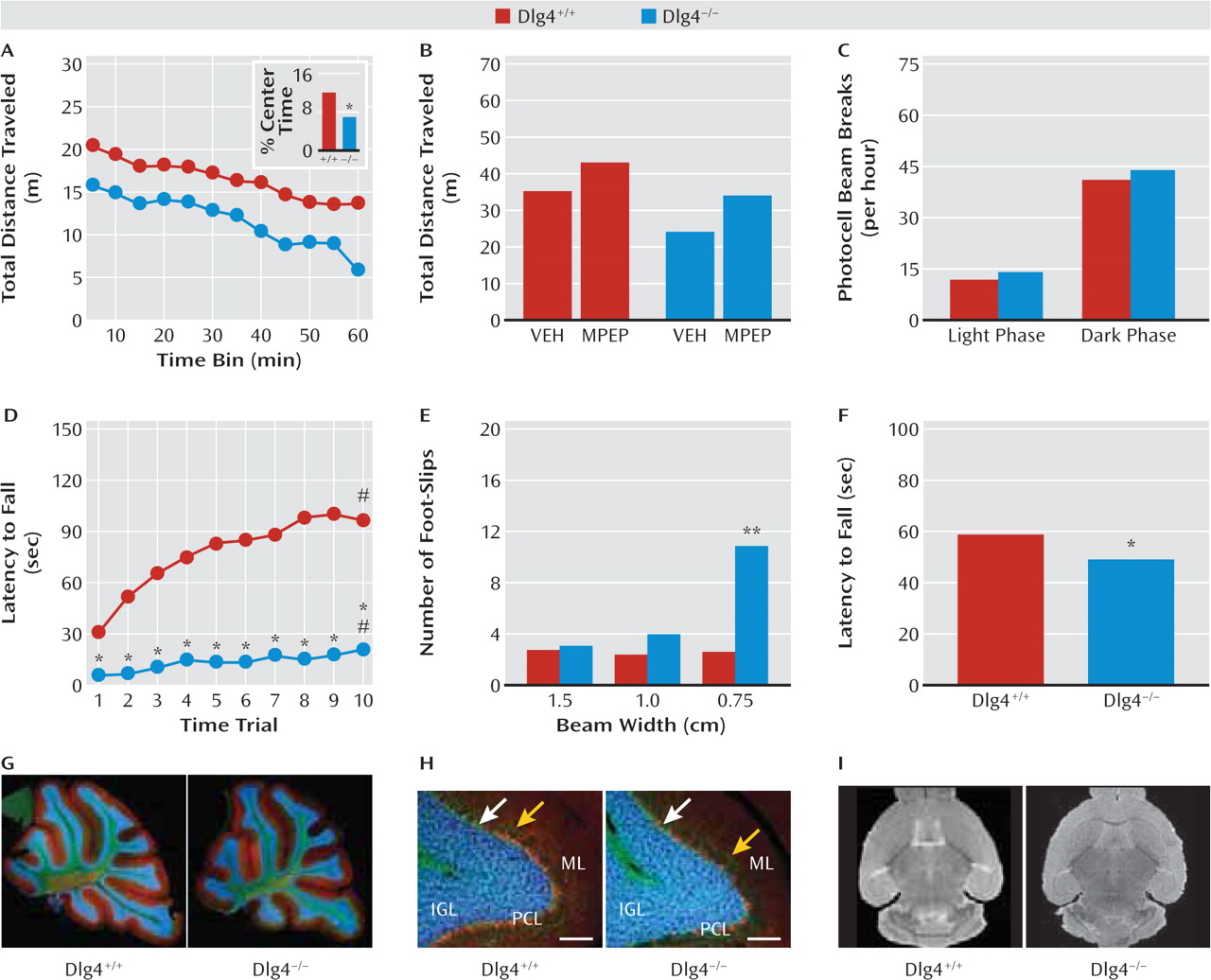
In addition to repetitive motor actions, autism typically presents with various motor deficits, including clumsiness; poor balance and coordination; unusual gait patterns, such as toe-stepping; hypotonia; and either locomotor hyperactivity or motor retardation (
21). A recent meta-analysis concluded that motor incoordination should be considered a cardinal feature of autism (
30). Consistent with such problems in motor coordination,
DLG4−/− displayed impaired performance on the accelerating rotarod test, as well as poor balance and gait on the narrowest balance beam and mild hypotonia on an inverted hang test. Similar deficits are seen in various glutamate signaling molecule mutants, such as
lurcher and
Grid2−/−, due to cerebellar developmental malformation (
31). However, our nonquantitative histological analysis did not reveal gross abnormalities in cerebellar lamination or foliation in
DLG4−/−. Gross brain morphology and volume, as measured by structural MRI, also appeared normal. While these data clearly demonstrate that
DLG4−/− phenocopy another major feature of autism, this appears to reflect yet-to-be-determined subcellular or physiological anomalies in the cerebellum or other brain regions controlling motor functions (e.g., striatum, motor cortex) rather than any major anatomical aberration.
Also within the motor domain,
DLG4−/− had reduced loco-motor activity in a novel open field. This was associated with increased center avoidance, suggesting that the hypo-activity could reflect anxiety-driven suppression of exploration rather than a more generalized locomotor retardation that might be related to the motor incoordination. Affirming this interpretation,
DLG4−/− showed normal levels of activity in the nonstressful homecage environment and normal behavior in another locomotion-based but putatively less stressful anxiety-related test: light/dark exploration. Moreover, treatment with the anxiolytic drug MPEP (
32) was sufficient to normalize open-field hypoactivity in
DLG4−/− to levels of untreated
DLG4+/+—an effect that would be difficult to explain if the mutants were simply motor compromised. We also tested
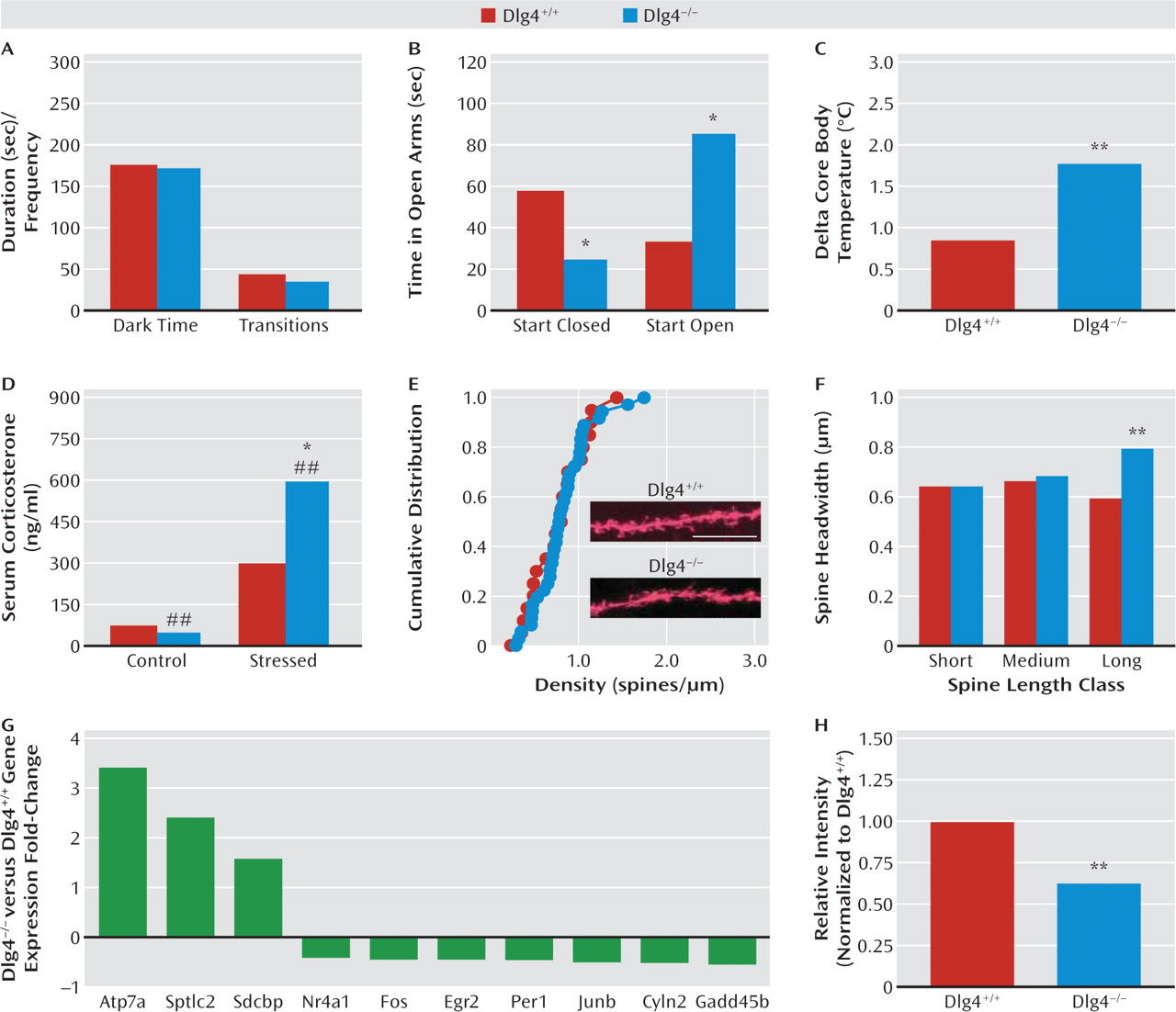
DLG4−/− in the elevated plus-maze, but behavior here was difficult to interpret because of a strong influence of start location. However, physiologic and neuroendocrine responses to stress were exaggerated in
DLG4−/−. Taken together, this profile points to exaggerated stress reactivity and a task-dependent increase in anxiety-like behavior in
DLG4−/− and provides a further parallel with the emotional dysregulation, oftentimes increased anxiety (
21), found in most neurodevelopmental disorders.
While the neural basis of these behavioral abnormalities remains to be elucidated, we found that
DLG4−/− had a subtle and specific alteration in the morphology of dendritic spines on basolateral amygdala neurons— a key neural locus mediating emotional and social behavior. Specifically, the heads of a subset of relatively long synaptic spines on basolateral amygdala neurons were enlarged in
DLG4−/−. This change was not associated with an alteration in spine density, nor in the spine density or morphology of spines in a prefrontal region that interacts with the basolateral amygdala to regulate emotion, the anterior cingulate cortex (
33). It would be premature at this time to draw clear links between these changes and the behavioral abnormalities exhibited by
DLG4−/−. It is an intriguing observation, however, considering that dendritic spine dysmorphology is increasingly linked to neurodevelopmental disorders (
34) and given evidence that
DLG4 regulates microtubule dynamics and motor proteins, such as dynein, that support the cytoskeletal machinery modulating dendritic spine morphology (
14,
35). It is also worth noting that spine head size correlates with synaptic strengthening (
36), particularly in the context of previous work demonstrating that
DLG4−/− have enhanced long-term potentiation at hippo-campal and corticostriatal glutamate synapses, coupled with impaired spatial reference memory and psycho-stimulant sensitization (
20,
37).
Providing some initial insight into molecular factors that might contribute to the spine morphology and/or behavioral disturbances caused by
DLG4 deletion, gene expression analysis revealed forebrain expression in a suite of synaptic genes, including genes associated with autism (
GADD45B,
Per1) and Menkes' disease (
Atp7a) (
38,
39). Another striking change was in
Cyln2 (cytoplasmic linker protein of 115 kDa, CLIP-115), which was decreased almost 50% at both the mRNA and the protein level. The human
CYLN2 gene is hemideleted within a microdeletion at chromosome 7q11.23 in Williams' syndrome (
23,
40–42). In fact, along with
LIMK1 and
GTF2I,
CYLN2 loss is a principal candidate for two unusual features of this disorder—hypersociability coupled with heightened anxiety to nonsocial stimuli (
23). While we do not claim that loss of
Cyln2 renders
DLG4−/− a model of Williams' syndrome, there are interesting parallels with the increased social and anxiety-related phenotype we observed in these mice. In another parallel, Williams' syndrome typically presents with motor incoordination manifested as abnormal gait and impaired stair stepping.
DLG4 is not located within the Williams' syndrome region or the orthologous region on mouse chromo-some 5. Previous studies have found that targeted heterozygous deletion of
Cyln2 impaired motor and fear-related behaviors (
43), while deletion of
Cyln2 along with proximally neighboring Williams' syndrome genes produced hypersociability (in choice, not free interaction tests), heightened anxiety-like behavior (in the novel open-field test, not in the light/dark test), and motor coordination deficits (
44). These similarities to the
DLG4−/− profile suggest a heuristic model in which
DLG4−/− deletion, via transacting epistatic or molecular interaction, alters
Cyln2 expression to drive, at least in part, the behavioral and possibly the morphological disturbances displayed by
DLG4−/−. Since
Cyln2 is thought to support proper synaptic clustering of proteins by bridging microtubules and protein complexes (
43), functional interactions with
DLG4 would not be unexpected. However, to our knowledge, this has not been directly demonstrated and remains a major question.
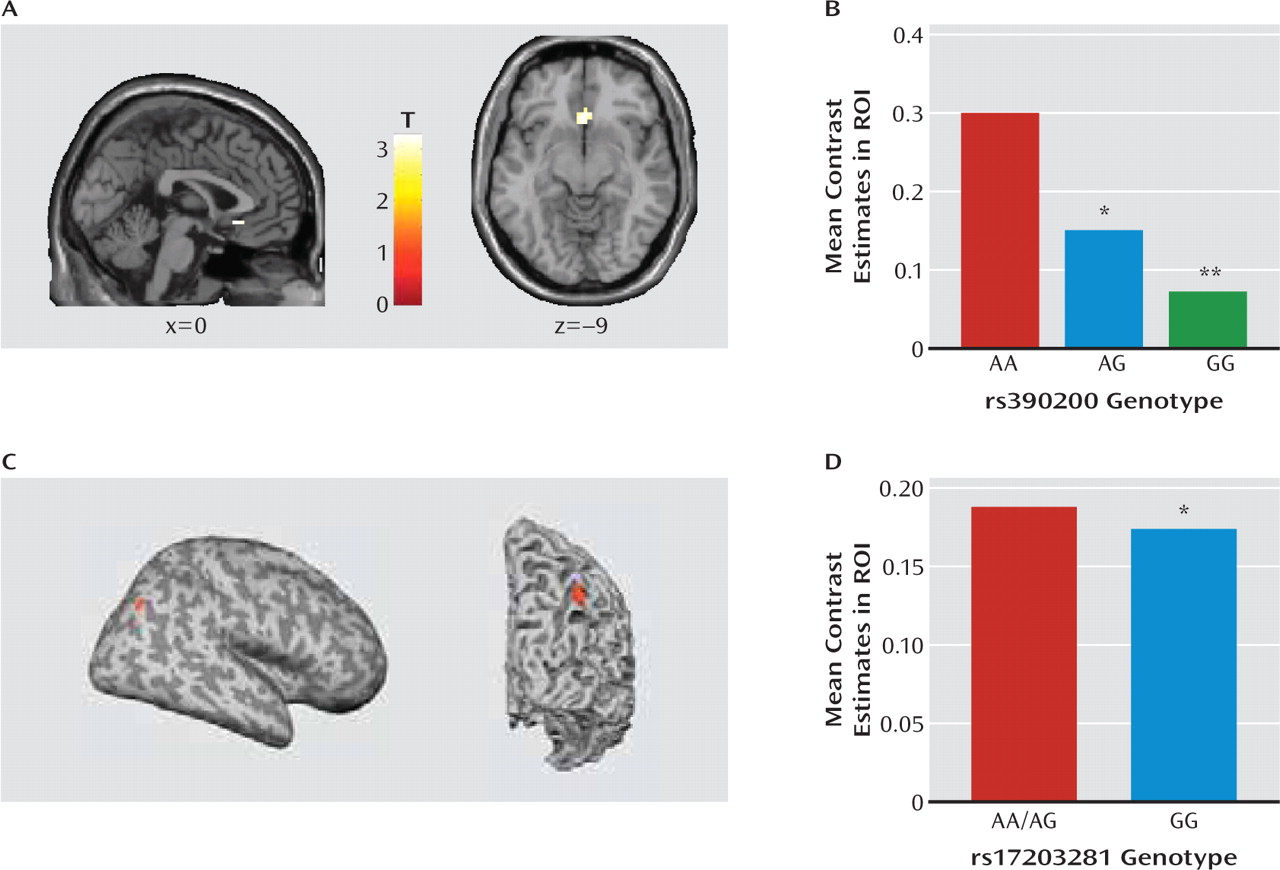
Extending these basic research findings, we provide preliminary evidence that variation in the human
DLG4 gene is associated with key neural Williams' syndrome endophenotypes. First, in a population of unaffected individuals, those carrying the G allele of
rs390200 had lesser subgenual anterior cingulate cortex-amygdala connectivity than homozygous A allele carriers during a threatening faces task (
25), mimicking the profile associated with the anxiety and hypersocial phenotype of Williams' syndrome (
23). Reduced cingulate-amygdala connectivity has also been linked to harm avoidance, an anxiety-related personality variable, in healthy individuals studied with the same task as employed here (
45). Second, homozygote carriers of the rs17203281 G allele had lower volume near the right intraparietal sulcus than A allele carriers, phenocopying the best-replicated structural abnormality in Williams' syndrome (
23). Although the functionality of these two SNPs remains to be determined, these data establish a link between
DLG4 variation and both functional and structural intermediate neural Williams' syndrome phenotypes. This finding raises some interesting questions for future work, such as whether these variants are present in Williams' syndrome patients and interact with the microdeletion to affect the clinical phenotype.