Over the past 20 years, substantial evidence has accumulated that structural variations of the human genome, especially copy number variations, are important causal factors for schizophrenia and related disorders (
1). Identification of associated copy number variations is shining new light on mechanisms of illness and on the genetically related spectrum of disorders (
2) that may include not only bipolar disorder but also autism, mental retardation, and epilepsy (
1). In this issue of the
Journal, Ingason et al. (
3) provide further data to suggest that neuropsychiatric conditions previously believed to be unconnected may share causal factors related to structural changes not visible on standard karyotype. Moreover, the parent of origin of the copy number variation in question, a duplication involving the proximal long (q) arm of chromosome 15, appears to be important.
Large, rare copy number variations that recur in unrelated individuals, including deletions (losses) and duplications (gains), are associated with genomic disorders (
4). Those with good evidence for expression of schizophrenia include deletions of the 22q11.2, 1q21.1, and 15q13.3 regions. Collectively, up to 2% of patients with schizophrenia may have such copy number variations (
1,
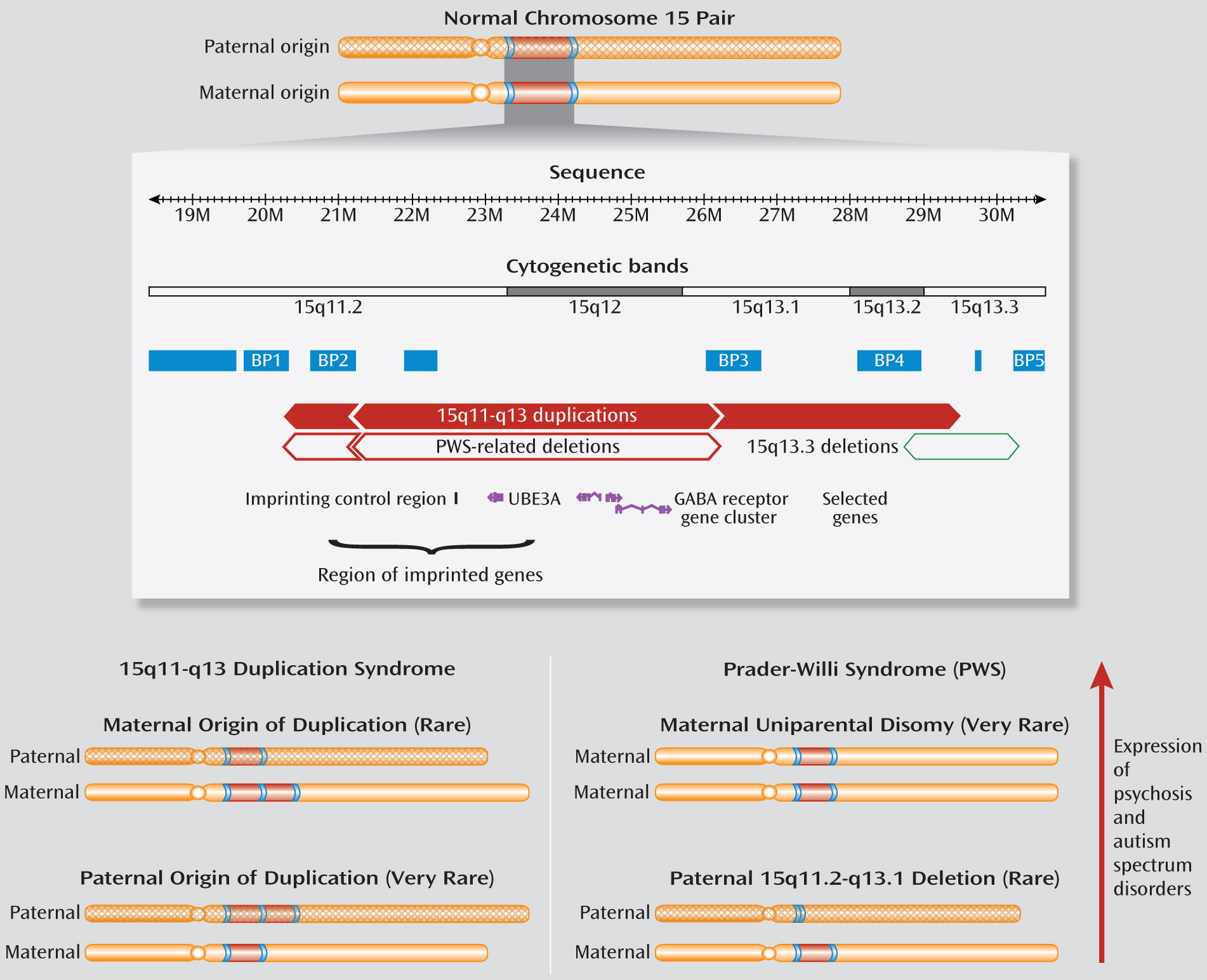
5). As for these copy number variations and those at other loci, repeated DNA sequences in the 15q11.2-q13.3 region, including several segmental duplications (
Figure 1), make this region prone to recurrent copy number variations (
4). Autism and mental retardation were the first conditions associated with excess copies of this region and the related syndrome (MIM 608636). Indeed, 15q11-q13 duplications are the most common anomaly in autism, accounting for 0.5%–3% of patients (
6).
What makes the copy number variations in this region special is that, in addition to the genomic structure, the parent of origin influences expression. Most duplications associated with autism are of maternal origin (
6). Also, this locus contains an imprinting control region (
Figure 1), with different methylation patterns of maternal and paternal alleles, which affects expression of several genes essential for neurodevelopment (
6). Prader-Willi syndrome (MIM 176270), featuring developmental delay, short stature, obesity, and hyperphagia, involves this imprinting mechanism. Adolescents and adults with Prader-Willi syndrome are at elevated risk of developing psychotic illness, usually depressive in nature. Prevalence of psychosis is significantly higher in those with maternal uniparental disomy (two maternal copies) than with a 15q11-q13 deletion of the paternal chromosome (
6,
7). This suggests that excess maternal copies of the 15q11-q13 region may play a more important role than absence of the paternal copy in increasing risk for psychotic illness (
6) (
Figure 1).
These findings set the stage for the Ingason et al. study reporting five patients with schizophrenia and 15q11-q13 duplications of maternal origin (
3). The Danish group found that one of 22 patients with childhood-onset schizophrenia and mental retardation had a 15q11-q13 duplication and enlisted other researchers with copy number variation data from several thousand individuals with schizophrenia, most of northern European descent, including three with previously published 15q11-q13 duplications (
3). Genome-wide copy number variation data from the Icelandic cohort are not publicly available, and thus eight other duplications appear for the first time (
3). The fact that this is one of the few imprinted regions in the human genome allowed the authors to determine the parental origin of the duplication copy number variations in the absence of parental DNA (
3), thus circumventing a common problem in studying adult-onset disorders. Their results support the likelihood that the phenotype of these duplications includes schizophrenia and that parental origin may influence this expression (
3), as it does for the associated autism phenotype (
6).
There were no 15q11-q13 duplications of either parental origin in any of thousands of comparison samples except those from Iceland (
3), suggesting that these copy number variations are very rare in the general population. Expression of the reported duplications included schizophrenia, bipolar disorder, and autism in probands as well as bipolar disorder in one mother. Comparison samples with the duplication had limited phenotypic data. Three of the five patients with schizophrenia had dual diagnosis with mental retardation or autism (two had unknown status). Available data suggest that the number of copies but not the length of the copy number variation may influence severity of expression (
6). Further studies of probands and relatives will be essential to delineate penetrance and variable expression of the phenotype as well as additional factors likely to be involved in expression. Smaller duplications that are associated with schizophrenia may help pinpoint the most plausible candidate gene(s) involved.
The authors list several limitations of their study (
6). Some are common to many recent copy number variation studies, including sampling biases and the incapability of returning to participants (
1). The latter compromised the ability to investigate the broader expression profile and to determine whether the duplications arose de novo or were inherited and cosegregated with illness in families. In addition, only a single algorithm was used to identify copy number variations (
1), and the prevalence of 15q11-q13 duplications in a catchment population of schizophrenia remains unknown. The limitation most would raise, however, is the small number of affected individuals.
Why are these findings important despite their apparent rarity? From a mechanistic standpoint, both the parent-of-origin effects and structural change of this chromosomal region may provide clues to the genes involved in and pathogenesis related to schizophrenia. Ingason et al. suggest that overexpression of maternally expressed imprinted genes, such as
UBE3A, could cause schizophrenia (
3), as proposed for autism (
6). Parent-of-origin effects are not restricted to classic imprinting mechanisms, however, and other genes may be involved. Normally, for example, the GABA
A receptor subunit genes
GABRB3,
GABRA5, and
GABRG3 (
Figure 1) are expressed equally from maternal and paternal alleles in the frontal cortex (
6,
8). There is genetic evidence implicating
GABRB3 in autism, including association of a rare coding sequence variant when it is maternally transmitted (
9). Importantly, dosage imbalance in the 15q11.2-q13.3 region can disrupt normal parental homologue pairing, DNA methylation patterns, and gene expression in this region during development of neurons (
6,
10). This process of homologue pairing of chromosome 15 requires binding of the MECP2-methylated DNA binding protein to sites across the 15q11.2-q13.3 region (
6,
8). Recent studies implicate fascinating epigenetic mechanisms involving this product of the
MECP2 gene, mutations of which cause Rett syndrome (MIM 312750). MECP2 deficiency has been shown to reduce expression of
GABRB3 as well as
UBE3A (
8,
10). Studies of gene expression in brain tissue unrelated to 15q11-q13 anomalies suggest the tantalizing possibility of broader applicability (
6,
10). Mouse- and human-induced pluripotent stem cell models may help further our understanding of a possible shared molecular pathway for autism and schizophrenia (
2).
Clinically, what do the Ingason et al. findings suggest? There is now a general recommendation for clinical genetic testing using genome-wide microarrays for all individuals with autism, mental retardation, and/or multiple congenital anomalies (
11). Thus, dual-diagnosis patients with schizophrenia and mental retardation, a traditionally understudied group, should be the first to receive such state-of-the-art investigations. This would include several of the study participants (
3). The proportion of patients with 15q11-q13 duplications and schizophrenia without other developmental conditions, thus not meeting these clinical criteria (
11), is unknown. Nonetheless, individuals with this genomic disorder deserve genetic counseling, now informed by results of this study. Recommendations for other clinically relevant changes in management or anticipatory care, such as those available for 22q11.2 deletion syndrome (
1,
12), await further data. Studies of rare copy number variations, like that by Ingason et al., can, however, bring us another step closer to understanding the variable expression that is the norm in genetic diseases and a step toward personalized medicine for patients with schizophrenia (
1).