An association between Alzheimer's disease and major depressive disorder has been reported in some studies, suggesting that depression could be considered either a risk factor for or a prodromal condition of Alzheimer's disease (
1–
7). In a meta-analysis of studies of depression and dementia, Jorm (
1) concluded that depressed individuals are, on average, nearly twice as likely to develop dementia, often in the form of Alzheimer's disease, compared with nondepressed comparison subjects. Similarly, depression was reported to be significantly associated with a higher rate of Alzheimer's disease in a population-based case-control study (
2). Multiple studies using a range of methods have generally strengthened the notion that major depression is a risk factor for Alzheimer's disease, even when it occurs earlier in life (
3–
7). However, there are exceptions (e.g., references
8,
9), and the presence of conflicting results suggests that there is heterogeneity among individuals with major depression with respect to the risk of Alzheimer's disease and that multiple pathological processes may be at play.
A potential link between major depression and Alzheimer's disease involves the role of amyloid beta in the brain. Disturbances in amyloid beta may be the earliest sign of Alzheimer's disease (
10). There are numerous amyloid beta peptide species, with the major isoforms consisting of two amino acid peptide fragments: 1–40 and 1–42 amino acid peptides. Amyloid beta peptides are a physiological product of the amyloid beta protein precursor through beta and gamma secretase cleavage (
11). Importantly, neuritic plaques, which are widespread in parenchymal brain tissue, are one of the neuropathological hallmarks of all forms of Alzheimer's disease (
12). Amyloid beta 42 in particular is known to be deposited early in plaques (
13) and is believed to be the initial trigger in Alzheimer's pathogenesis. CSF amyloid beta 42 is now considered a biomarker of Alzheimer's disease, and its levels appear to inversely reflect brain amyloid beta deposition, as demonstrated by in vivo studies using amyloid tracers, such as Pittsburgh compound B (
14). Consistent with these findings, lower CSF concentrations of amyloid beta 42 have been observed in individuals with Alzheimer's disease and mild cognitive impairment relative to comparison subjects (
15). Other important CSF biomarkers of Alzheimer's disease are levels of total tau protein, a marker of neuronal degeneration, and levels of hyperphosphorylated tau protein, a marker of neurofibrillary tangles. Both total and phosphorylated tau protein CSF levels are reported to be greater in individuals with Alzheimer's disease than in comparison subjects (
16).
Several lines of evidence suggest that amyloid beta disturbances may also be associated with major depressive disorder and depressive symptoms. Results from preclinical research, including primate studies, have associated various risk factors for depression with increased soluble amyloid beta production in the brain and increased amyloid plaques; among them are acute and chronic stress, glucocorticoid administration, sleep deprivation, and increased levels of corticotropin-releasing factor and cortisol secretion (
17–
19). Furthermore, it has been reported that when injected into the cerebral ventricles in rodents, amyloid beta 42 induces depression (
20). Lastly, several researchers have reported plasma amyloid beta 42 disturbances in humans, although the results have been inconsistent, with some depressed individuals having lower (e.g., reference
21) or higher (e.g., reference
22) amyloid beta 42 levels than comparison subjects. A major drawback of these studies is that plasma concentrations of amyloid beta 42 typically are not correlated with CSF or brain concentrations and are not considered a reliable marker of Alzheimer's disease pathology (
23).
CSF levels of amyloid beta and tau protein have also been studied in conjunction with major depression in elderly individuals. Gudmundsson et al. (
24,
25) compared elderly women (exclusively) with major depressive disorder with age- and sex-matched comparison subjects without depressive symptoms. Neither group had dementia, and the depressed group had higher CSF levels of amyloid beta 42 than comparison subjects. The exact significance of this finding is unclear but may correspond to increased amyloid beta production in the brain, increased amyloid beta clearance, or decreased amyloid beta brain deposition in individuals with major depression relative to comparison subjects with no depressive symptoms. In order to isolate the reasons behind increased amyloid beta 42 levels, researchers have employed in vivo positron emission tomography (PET) technology with ligands, such as Pittsburgh compound B to detect amyloid beta and [
18F]FDDNP (2-(1-{6-[(2-[fluorine-18]fluoroethyl)(methyl)amino]-2-naphthyl}ethylidene)malononitrile) to detect both amyloid beta and tau neurofibrillary tangles. Studies using this technology have revealed increased amyloid beta deposition in the brain in individuals with depression relative to nondepressed comparison subjects (
26). In particular, increased brain amyloid beta and tau protein binding were observed in currently depressed individuals with major depressive disorder and no mild cognitive impairment, relative to comparison subjects, in recent PET studies using Pittsburgh compound B (
27) and [
18F]FDDNP (
28).
Collectively, these results suggest that amyloid beta-related neurobiological mechanisms may play a role not only in Alzheimer's disease but also in late-life major depression. However, to our knowledge, the only studies to date to investigate CSF markers of Alzheimer's disease, such as amyloid beta and tau proteins, in cognitively intact elderly individuals with major depressive disorder examined women exclusively (
24,
25). The purpose of our study was to investigate CSF markers of Alzheimer's disease in cognitively intact elderly men and women alike. We predicted that, in line with previous findings from PET studies, those with major depressive disorder, regardless of gender, would have lower CSF levels of amyloid beta 42 relative to comparison subjects, indicating higher brain amyloid beta deposition, as seen in individuals with Alzheimer's disease and mild cognitive impairment. In contrast, we expected no significant differences in CSF levels of amyloid beta 40, total tau, or phosphorylated tau across groups, which is consistent with results reported previously by Gudmundsson et al. (
24) and Buerger et al. (
29).
In some studies, CSF isoprostane levels, considered to be a biomarker of oxidative stress (
30), which is characteristic of inflammation (
31), have been associated with CSF levels of amyloid beta 42 (
32). Both oxidative stress and inflammatory mechanisms have also been implicated in depression, and higher plasma isoprostane levels have been observed in elderly depressed patients relative to healthy comparison subjects (
33). Consequently, we also incorporated a measure of oxidative stress (F2-isoprostanes) as part of the present study.
Finally, three neuropsychological tests were administered to all participants: the Buschke Selective Reminding Test (
34), which measures memory performance; the Trail-Making Test, parts A and B (
35), which measures attentional and psychomotor performance skills; and the category fluency test (
36), which measures spontaneous verbal generative ability. These tests assess primary cognitive domains and were used to determine whether any cognitive differences were observable across groups.
Method
Participants
This study was approved by the institutional review boards of the Nathan Kline Institute for Psychiatric Research and the New York University School of Medicine. Participants were volunteers who responded to advertisements in local newspapers and flyers or were recruited from our currently active Memory Education and Research Initiative Program. All participants provided formal consent prior to being examined and were compensated up to $450.00. A total of 133 participants completed the baseline evaluation, and 51 of these took part in the optional lumbar puncture procedure. Of the 51 participants who had lumbar puncture, three were excluded because of evidence in their MRI scans of confluent deep or periventricular white matter hyperintensities, defined as one or more hyperintense lesions measuring at least 10 mm in any direction. One individual was excluded because of a Mini-Mental State Examination (MMSE) score below 28. Of the 47 remaining participants, 28 were diagnosed with major depressive disorder by a board-certified psychiatrist based on clinical evaluation and the Structured Clinical Interview for DSM-IV Axis I Disorders (SCID), leaving 19 comparison subjects. Of the 28 individuals with major depressive disorder, 21 (75%) had recurrent episodes. A Clinical Dementia Rating scale score was compiled for 33 participants, and none of these had a score above 0.
Table 1 summarizes the demographic and clinical characteristics of the study participants.
Amyloid Beta and Tau Protein Determination
We analyzed CSF levels of amyloid beta 1–40 and 1–42 with electrochemiluminescence technology using the MS6000 Human Ab Ultra-Sensitive Kit (Meso Scale Discovery, Gaithersburg, Md.). We determined the total tau concentration in CSF using a sandwich enzyme-linked immunosorbent assay (ELISA) (Innotest hTAU-Ag, Innogenetics, Ghent, Belgium) specifically constructed to measure all tau isoforms, irrespective of phosphorylation status. Tau protein phosphorylated at threonine 181 was measured using a sandwich ELISA method (Innotest Phospho-Tau [181P], Innogenetics).
Isoprostane Determination
First, CSF samples underwent extraction by use of a C18 cartridge and then were purified by thin-layer chromatography. Next, F2-isoprostane levels were assayed by a specific and sensitive sandwich ELISA method, previously described by Zhao et al. (
30).
Apolipoprotein E Genotyping
We collected 5 ml of whole blood for apolipoprotein E (APOE) genotyping. The blood samples were transferred to a cryogenic tube and stored at –80°C until genotyping. Genomic DNA samples were prepared from whole blood. We determined APOE genotype using standard methods of polymerase chain reaction (PCR)-restriction fragment length polymorphism, with Hhal digestion of an APOE genomic PCR product spanning the polymorphic (cysteine-arginine) sites at codons 112 and 158, analyzed using 12% polyacrylamide gel electrophoresis.
Design and Analysis
Our objective was to test whether major depressive disorder was associated with CSF levels of amyloid beta, tau protein, and isoprostanes. First, t and chi-square tests were used to compare the major depression and comparison groups with respect to dimensions not associated with depression (i.e., MMSE score, years of education, body mass index, age, incidence of diabetes, gender,
APOE status, and reported family history of Alzheimer's disease). Second, since the only previous studies of CSF levels of amyloid beta in relationship with major depression (
24,
25) examined only women, CSF levels of amyloid beta 42 were analyzed with a two-factor analysis of variance (ANOVA) that crossed major depression status with gender. Finally, t tests were used to compare the two diagnostic groups with respect to all remaining CSF variables. All tests were two-tailed, and statistical significance was established at an alpha of 0.05. All analyses were conducted using SPSS 15.0 (SPSS, Inc., Chicago).
Procedure
The study was conducted over four visits, generally 1 week apart. The first three visits were conducted at the Nathan Kline Institute for Psychiatric Research and the Clinical and Translational Science Institute, New York University Langone Medical Center. During the first visit, for the purpose of obtaining informed consent, study procedures were explained and participants were informed of their rights. Participants' medical and psychiatric histories, including family history of Alzheimer's disease, were obtained, and their vital signs were measured. Participants also underwent a psychiatric evaluation, and their global cognitive status was assessed using the MMSE. When a collateral informant was available (N=33), the Clinical Dementia Rating scale was also completed. Additionally, the Hamilton Depression Rating Scale (HAM-D) was administered to rate the severity of current depressive symptoms. Blood was drawn for routine medical testing and
APOE genotyping. During the second visit, participants underwent an MRI scan of the head to quantify the magnitude of vascular brain pathology. During the third visit, they underwent a comprehensive neuropsychological assessment, which included the Buschke Selective Reminding Test (
34), the Trail-Making Test, parts A and B (
35), and the category fluency test (
36). The Buschke Selective Reminding Test consists of a list of 16 unrelated nouns, presented orally at a rate of 2 seconds each. The subject is asked to recall as many words as possible and to indicate when no more words can be recalled. Seven presentation and recall trials of the same list of nouns are given in immediate succession. The subject is asked to recall the entire list on each trial, but after the initial presentation, he or she is presented with only those words that were not recalled on the trial immediately preceding the current trial. Finally, after an interval of 20 minutes, a delayed recall test is also given, in which the participant is asked to free recall as many words from the list as possible. In Trails A, participants are required to draw a series of lines connecting 25 numbered circles (after practice with five numbered circles). The time it takes to complete the test is taken as the measure of performance. Trails B is identical to Trails A except that the circles contain both numbers and letters, and an alternating sequence must be followed (i.e., 1-A-2-B-3-C). For the category fluency test, participants are given a semantic category (e.g., animals) and are asked to produce as many category words as possible in 60 seconds.
During the fourth visit, a lumbar puncture was performed by a neuroradiologist under guided fluoroscopy. Participants were asked to fast overnight (12 hours) prior to the procedure, which was performed between 9:00 a.m. and 10:00 a.m. A total of 15 ml of clear CSF was collected in three polypropylene tubes labeled “A” (first 5 ml), “B” (second 5 ml), and “C” (third 5 ml). The tubes were immediately placed on ice for a maximum of 1 hour until the samples were centrifuged at 4°C (at 1500 rpm) for 10 minutes. Then, aliquots of 0.25 ml were placed into 1.00-ml polypropylene cryogenic vials and put into Nunc eight-cell storage boxes (Nalge Nunc International, Rochester, N.Y.) at –80°C. All amyloid beta and tau determinations were performed from tube “C,” whereas isoprostanes were determined from tubes “A” and “B.”
Results
Aside from major depression status, the two study groups did not differ in any relevant clinical or demographic variable with the exception of the mean HAM-D score, which was significantly higher in the major depression group (
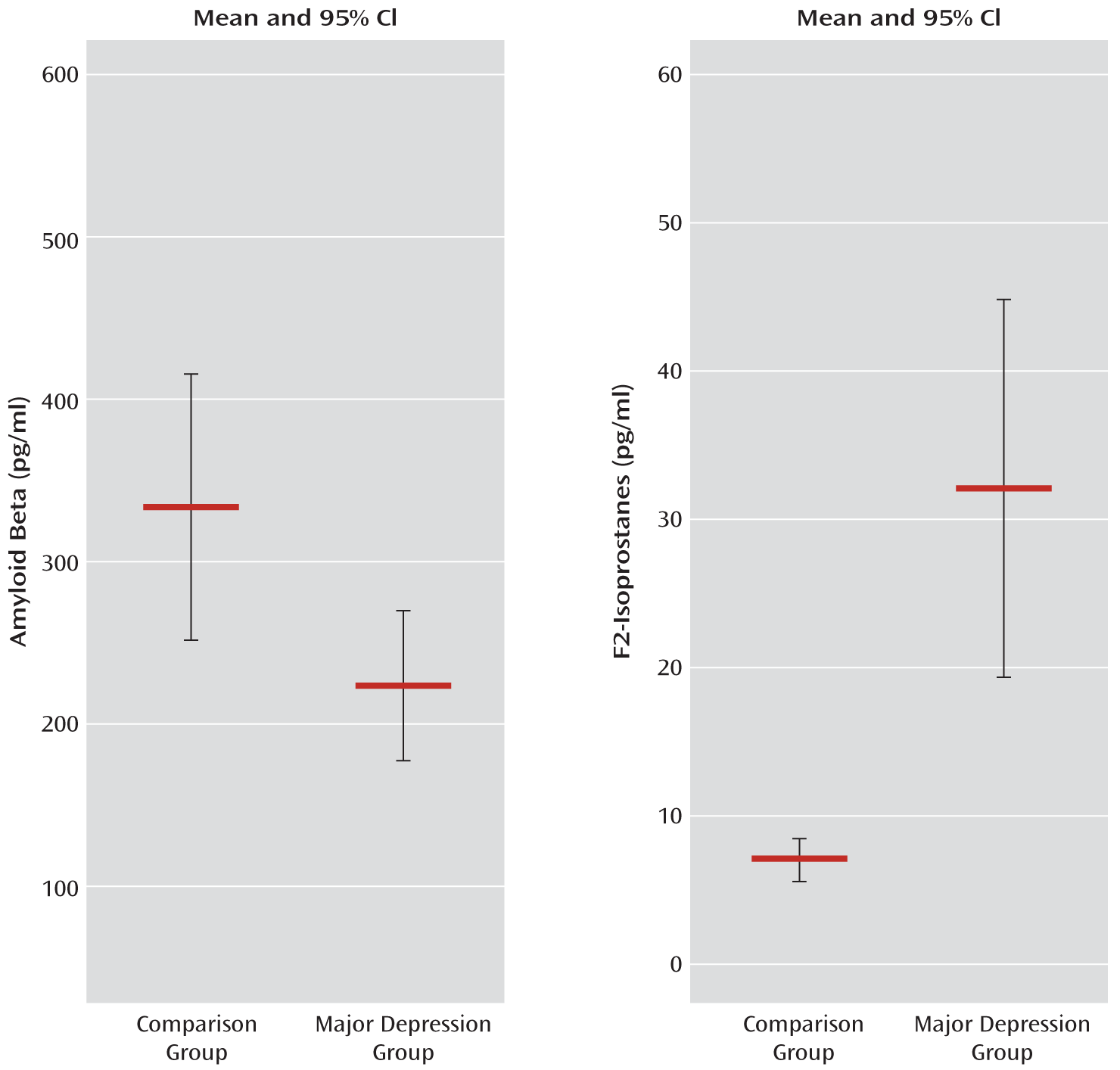
Table 1). Of note, the proportion of participants with a reported family history of Alzheimer's disease was slightly higher in the comparison group than in the major depression group. Although this difference fell short of statistical significance, it is still contrary to our hypothesis and thus cannot account for our findings. The results from a two-factor ANOVA of amyloid beta 42 levels indicated that there was no interaction between major depression status and gender. There was a main effect of major depression status (F=5.321, df=1, 43, p=0.03), indicating that individuals with major depression had lower amyloid beta 42 levels than comparison subjects, but there was no main effect of gender. Using t tests and excluding gender, we confirmed that amyloid beta 42 levels differed significantly across conditions (t=2.471, df=45, p=0.02) (
Figure 1), whereas levels of amyloid beta 40 fell short of statistical significance across conditions (
Table 2). In contrast, total and phosphorylated tau protein levels did not reach statistical significance across conditions (
Table 2). The difference in amyloid beta 42 levels was still present (t=2.067, df=29, p<0.05) when we excluded all individuals (from both groups) with the
APOE ε4 allele (N=16/47), who typically have lower CSF levels of amyloid beta 42. Another potential cause of lower levels of amyloid beta 42 is mild cognitive impairment as a result of early Alzheimer's disease. Although we did not specifically diagnose participants for mild cognitive impairment, we attempted to address this issue by operationally defining mild cognitive impairment as a delayed recall score that was 1.5 or more standard deviations below the mean score of the entire sample. Three participants met this threshold (two in the comparison group and one in the major depression group). With these three participants excluded, the two-factor ANOVA of amyloid beta 42 levels still yielded a main effect of major depression status (F=4.170, df=1, 40, p<0.05). Additionally, no interaction between major depressive disorder status and gender and no main effect of gender were observed after excluding these individuals, thus confirming that mild cognitive impairment was an unlikely cause for a major depression-related effect on amyloid beta 42 levels.
Similarly, other cognitive measures (Trails A and B, category fluency test) revealed no differences across groups, thus making it less likely that the major depression group had more cases of mild cognitive impairment than the comparison group.
Importantly, we also observed an inverse correlation between amyloid beta 42 levels and HAM-D scores, in both the whole sample (r=–0.415, p=0.004) and in the major depression group only (r=–0.390, p=0.04), indicating that lower levels of amyloid beta 42 were associated with greater symptom severity. Fifteen individuals with major depression were receiving antidepressant treatment at the time of testing, but no differences in amyloid beta 42 levels were observed within the major depression group as a function of antidepressant treatment (no antidepressants: mean=211.5 [SD=94.3]; antidepressants: mean=236.1 [SD=149.1]).
After removing three significant outliers, we observed that F2 isoprostane levels differed significantly across conditions (t=3.818, df=38, p=0.001), with much higher levels seen in the major depression group (N=27) than in the comparison group (N=13) (
Figure 1). These differences suggest that increased oxidative stress is associated with depression. Additionally, similar to previous reports in the literature, we observed that levels of amyloid beta 42 inversely correlated with F2-isoprostane levels (r=–0.331, p=0.04) when both individuals with major depression and comparison subjects were considered. This correlation was not significant when considering depressed individuals only, although this may be a consequence of lower statistical power.
Discussion
Our results confirm that elderly, cognitively intact individuals with major depressive disorder have reductions in CSF levels of amyloid beta 42 similar to individuals with Alzheimer's disease or mild cognitive impairment. This pattern is typically thought to reflect increased deposition of amyloid beta in the brain, consistent with PET results from studies using Pittsburgh compound B and [
18F]FDDNP (
26–
28). These reductions in amyloid beta 42 levels do not appear to be associated with gender (
24,
25). Moreover, unlike in individuals with Alzheimer's disease and mild cognitive impairment, differences in CSF tau protein levels were not observed in individuals with major depression in our study. However, the development of tau pathology in Alzheimer's disease is known to be a downstream phenomenon relative to amyloid beta deposition. Thus, longitudinal studies are needed to determine whether cognitively intact individuals with major depression might eventually develop increased tau levels, which would be expected in those with prodromal Alzheimer's disease.
Our results are in contrast with the only previous study that examined CSF levels of amyloid beta 42 in individuals with major depression and comparison subjects (
24). The authors of the previous study reported that CSF levels of amyloid beta 42 were higher in 11 elderly women with major depressive disorder and no dementia compared with 65 age-matched, cognitively intact healthy comparison subjects. Although we cannot explain this discrepancy with certainty, one possibility is that there was a difference in the type of depressed individuals included. The depressed volunteers included in our study were mostly individuals with recurrent/chronic symptoms (75%), whereas no exact information on this characteristic was provided in the previous study (
24). Thus, it is possible that a majority of the 11 depressed patients in the previous study represented acute cases, which is not uncommon in a population-based study. These cases could have involved elevations in CSF levels of soluble amyloid beta 42 prior to the formation of senile plaques, after which decreased efflux from brain interstitial fluid and a reduction in amyloid beta 42 levels would be observed.
In addition to a decrease in amyloid beta 42 levels across groups, we also observed a similar effect in levels of amyloid beta 40 that approached statistical significance. Although the interpretation of this finding requires caution, unlike amyloid beta 42, amyloid beta 40 is known to be preferentially deposited in vessel walls. Thus, it is possible that a reduction in CSF levels of amyloid beta 40 may reflect increased deposition of this peptide in the cerebral blood vessels of individuals with major depression relative to nondepressed comparison subjects. If confirmed, this finding could have implications for our understanding of neurobiological factors that may contribute to the well-documented association between depression and cerebrovascular disease (
37).
A decrease in CSF levels of amyloid beta 42 (and amyloid beta 40) not accompanied by concomitant increases in total and phosphorylated tau protein levels, as occurs in Alzheimer's disease, might also indicate decreased production of amyloid beta, not just increased deposition. Although the physiological function of amyloid beta is unknown, reports indicate that lower production of this peptide may be associated with lower neuronal and synaptic activity (
38). Therefore, the observation of lower CSF levels of amyloid beta 42 in major depression might reflect decreased brain activity in depressed individuals relative to nondepressed comparison subjects.
We observed that individuals with depression also had indications of increased oxidative stress, as measured by levels of F2-isoprostanes. Isoprostanes are a product of reactive oxygen species-induced lipid peroxidation and serve as biomarkers of oxidative stress (
30), which has been associated with depression (
33). Importantly, the amyloid beta 42 level was found to be inversely correlated with F2-isoprostane levels, suggesting that more oxidative stress was present when levels of amyloid beta 42 were lower. Moreover, we also observed that lower amyloid beta 42 levels were associated with more depressive symptoms. Collectively, these findings are consistent with a large body of preclinical and clinical data suggesting that higher brain amyloid beta deposits have deleterious effects on brain structure and function. In particular, since the severity of depressive symptoms is linked with lower CSF levels of amyloid beta 42, neuronal and synaptic disruption may be particularly relevant in areas associated with mood regulation, such as the medial prefrontal cortex and medial temporal lobe structures, including the amygdala. These regions are part of the default mode network, which has been shown to be a premier site of the distribution of amyloid beta plaques (
39).
Fifteen participants in our major depression group were receiving treatment with antidepressant medication at the time of testing, but we observed no within-group difference as a function of antidepressant treatment. Collectively, our findings suggest that conventional antidepressant therapy may not be effective in correcting brain amyloid beta disturbances. This inefficacy is not surprising because all currently available antidepressants appear to target solely the monoaminergic system and related receptors (
40). However, a large number of individuals do not respond to monoamine-based antidepressants or their combinations, and those who do often relapse, even while complying with the therapeutic regimen. Consequently, there is a clear need for the identification of novel molecular candidate mechanisms that may underlie the pathogenesis of depression, with the aim of improving current therapeutic approaches to the treatment of major depression. The relationship between major depression and Alzheimer's disease biomarkers, as described in this study, suggests a common pathological process that, while not necessarily causative, may play a significant role in the development of both disorders.
Future research should consider whether our results might extend to a younger population (under age 60) or to individuals suffering from acute depression, rather than chronic depression as in our study. Moreover, a longitudinal study is warranted to explore how (or whether) CSF levels of amyloid beta 42 might change over time in individuals with major depressive disorder and whether the changes are associated with the emergence of progressive cognitive decline or brain structural and functional abnormalities.
Acknowledgments
The authors thank Dr. Leslie A. Saint-Louis for performing the lumbar punctures. The authors also thank Ms. Francesca M. Pomara for assistance in revising an earlier version of this article.