Schizophrenia is a complex psychiatric illness with interplay between environmental and genetic components. Among the neurotransmitter systems implicated in this illness, dopamine has been at the forefront of research for almost 50 years (
1–
4), and the singular target for the treatment of schizophrenia has been the dopamine receptor (D
2 subtype) (
1–
3). The development of atypical antipsychotics provided new therapeutic tools with fewer motor side effects but with more metabolic effects (
4–
6). The available therapeutic agents rarely induce recovery, suggesting that none are specific enough to address the complex pathophysiology of the disease.
One of the variable limitations of the available antipsychotic drugs is prolongation of the QT interval (long QT syndrome) and higher rates of sudden death that are potentially related to this side effect (
7). The QT prolongation effect results from blockade of the hERG1 potassium channel in the heart (
8,
9). hERG1 belongs to a subfamily of voltage-gated K
+ channels that plays a critical role in regulating excitability and repolarization in cardiac muscle and smooth muscle as well as in neurons (
10). The hERG1 channel is responsible for a delayed rectifier potassium current, a major component of cardiac repolarization (
10,
11). The hERG1 protein is encoded by the
KCNH2 gene on 7q36.1. Over 300 genetic variants in
KCNH2 have been identified that can either cause a congenital form of long QT syndrome or predispose an individual to an acquired long QT syndrome (
12,
13). However, blockade of hERG1 channels by antipsychotics may represent the most frequent cause of an acquired long QT syndrome (
14).
Using mRNA characterization techniques in postmortem human brains, Huffaker et al. (
15) identified a brain-selective and primate-specific variant of
KCNH2 (isoform 3.1) that regulates neuronal firing patterns. When compared with the full-length
KCNH2 (1A) isoform,
KCNH2 3.1 mRNA levels are similar in the brain but lower by three orders of magnitude in the heart. The
KCNH2 3.1 isoform has a novel 5′ exon extension and is lacking the PAS domain of the full-length protein that is critical for slow channel deactivation and repolarization. Meta-analysis of five independent clinical data sets showed an association between single-nucleotide polymorphisms (SNPs) in
KCNH2 and schizophrenia, and the risk-associated alleles predicted lower IQ and lower cognitive processing speed even in healthy subjects. The same risk-associated alleles also predicted greater
KCNH2 3.1 mRNA expression in the postmortem hippocampus of both schizophrenia patients and healthy subjects, implicating a molecular mechanism of the clinical associations. Moreover, the relative expression of the 3.1 isoform was 2.5 times higher in patients relative to control subjects, suggesting a pathophysiological role of
KCNH2 3.1 in the clinical state. The
KCNH2 3.1 isoform was shown to have unique physiological properties in neuronal culture, mediating fast neuronal spiking (
15).
Genome-wide association studies (GWAS) of schizophrenia have not found evidence of association with
KCNH2, but this may reflect the fact that current GWAS data are primarily from SNP chips lacking coverage of
KCNH2. Two recent targeted genetic studies have confirmed the association of the same SNPs and alleles in
KCNH2 with schizophrenia (
16,
17). While it is not known whether antipsychotics affect the brain-specific
KCNH2 3.1 channel as they do the full-length channel, the fact that antipsychotics have hERG1 effects and that
KCNH2 3.1 is genetically associated with schizophrenia at both the clinical and molecular levels raises the possibility that the therapeutic effects of antipsychotics may relate at least in part to changes in
KCNH2 3.1 activity.
In this study, we evaluated the impact of genetic variation in KCNH2 in two independent clinical trials of antipsychotic medications in patients with schizophrenia: a National Institute of Mental Health (NIMH) double blind, placebo-controlled inpatient crossover study, and the NIMH-sponsored multicenter Clinical Antipsychotic Trials in Intervention Effectiveness (CATIE) study. We tested the hypothesis that KCNH2 genotypes that have been shown to be related to risk for schizophrenia and to expression of isoform 3.1 of KCNH2 influence treatment response to antipsychotic drugs. The results of both independent trials support this hypothesis.
Method
Sample
Two independent cohorts of patients enrolled in antipsychotic treatment trials were included in this study.
NIMH cohort.
The first cohort consisted of 54 partially treatment-resistant patients with schizophrenia who were admitted to the Clinical Brain Disorders Branch schizophrenia inpatient research unit at the National Institutes of Health (NIH) Clinical Center between 1998 and 2010. All of these patients were self-reported Caucasians of European ancestry and were diagnosed with chronic schizophrenia using DSM-IV criteria. They volunteered to participate in a double-blind, placebo-controlled crossover study with standard doses of atypical antipsychotics including olanzapine, quetiapine, risperidone, ziprasidone, and aripiprazole. All patients, and a family member if necessary, provided informed consent for participation in the study, which was approved by the NIH institutional review board. The program typically enrolled patients who previously had adequate trials of standard antipsychotic medication interventions that were only partially effective in decreasing their symptoms. Upon admission, all patients underwent medical, neurological, and psychiatric evaluation lasting 4–12 weeks. If it was then concluded that the patient had significant medical problems, a history of violence, or suicidal behavior not identified during the prehospitalization screening or if the patient decided to withdraw consent, he or she could leave the hospital or participate in other protocols not involving placebo studies. The demographic characteristics of the patients are summarized in
Table 1.
All patients included in this study were taking atypical antipsychotics before their admission. After the initial evaluation period, patients were maintained on a standard dosage of one atypical antipsychotic, and all other medications were discontinued. The decision about which antipsychotic would be used in the blinded study was based on the best previous response, patient preference, and side effect profile. The patients then remained on the single antipsychotic in an open-label fashion for several weeks before transitioning to the coded trial medication and being tapered from their other medication over a period of 4 to 7 days. Subgroup 1 (N=35) underwent a sequence of 4 weeks of coded placebo followed by 4 weeks of a coded standard atypical antipsychotic. Subgroup 2 (N=19) underwent the inverse sequence (i.e., after a similar open medication and taper period, they received standard atypical antipsychotic treatment for 4 weeks followed by placebo for 4 weeks). Because of the highly supervised and structured nature of the NIMH inpatient environment, most patients tolerated the 4 weeks of placebo well. At the end of the coded double-blind protocol, the patients restarted an active medication for 6 or more weeks before they started another experimental protocol or until they were well enough to be discharged.
Weekly assessments with the Positive and Negative Syndrome Scale (PANSS;
18) were conducted independently by one of four trained research nurses. Two weeks before starting the double-blind protocol and up to 4 weeks after completing it, these evaluations were performed twice a week. The mean PANSS subscores in the NIMH study before the patients entered the placebo-controlled protocol were as follows: 14.25 for positive syndrome (SD=4.09), 15.69 for negative syndrome (SD=4.6), 29.58 for general psychopathology (SD=5.81), 8.48 for anergia (SD=2.23), 8.36 for thought disturbance (SD=3.61), 5.08 for activation (SD=1.31), 4.89 for paranoid/belligerence (SD=0.85), and 8.40 for depression (SD=2.18).
CATIE cohort.
The second cohort consisted of patients randomly assigned to one of five medications during phase 1/1A (first drug assigned) of the CATIE study (
19), an NIMH-sponsored multicenter clinical trial comparing the effectiveness of various antipsychotic drugs. The demographic characteristics of the patients in the CATIE cohort are summarized in
Table 2. Initially, patients received olanzapine, perphenazine, quetiapine, risperidone, or ziprasidone under randomized double-blind conditions, and follow-up occurred for 18 months, until treatment was discontinued, or until the patient switched to another antipsychotic (phase 2). Eligible patients were 18 to 65 years old, had received a diagnosis of schizophrenia based on DSM-IV criteria, and were able to take oral antipsychotic medication. The primary outcome measure in the CATIE sample was time to medication discontinuation, which occurred most often because of side effects or lack of efficacy, and PANSS scores (positive syndrome, negative syndrome, and general psychopathology) were included as a secondary efficacy outcome. Details on the CATIE methods have been published elsewhere (
19). To be consistent with the NIMH inpatient study and to reduce the effects of genetic heterogeneity, only Caucasian patients were included for analysis. Because of the outpatient and parallel design of this study, information about compliance based on drug clearance is an important factor for determining symptom change in participants of CATIE (
20). We therefore analyzed treatment response only from patients with drug clearance data (N=364) (
20). As an ancillary project to the CATIE study, blood samples were taken during study visits to measure antipsychotic drug concentrations. Data on the amount of the last dose of medication, time of last dose, and time of blood sample were also collected. This information was used with the drug concentration data to estimate drug clearance for each patient based on nonlinear mixed-effects modeling using NONMEM (version 5, level 1.1; Ellicott City, Md., GloboMax). We used drug clearance instead of plasma concentrations because it is a dose-independent and time-independent measure, which allows for comparison of drug exposure across all subjects, as described in detail elsewhere (
20,
21).
Genotyping
We genotyped three SNPs (rs3800779, rs748693, and rs1036145) in
KCNH2 that have been associated with schizophrenia and with expression of the novel
KCNH2 3.1 isoform in the postmortem brain (
15). Genotypes were determined using the 5′-exonuclease fluorescent Taqman assay (assay by design or assay on demand), and the allelic discrimination was read on an ABI 7900 SDS system (Applied Biosystems, Foster City, Calif.) (
22).
Analyses
Several statistical analyses based on the specifics of study design and outcome measurement were performed. The NIMH study used a randomized, double-blind, placebo-controlled crossover design in which PANSS scores were evaluated as a primary outcome. Since the PANSS was repeatedly administered over time in both the placebo and treatment arms of the study, with few instances of discontinuation due to clinical decompensation, we employed a general linear mixed-effect full model that included the main effects of treatment (antipsychotics compared with placebo) and genotype, and the interaction of genotype by treatment, while controlling for duration of illness, sex, and age. In the CATIE study (cohort 2), time to discontinuation of medication was considered the primary outcome, and PANSS scores were a secondary outcome measure. However, to replicate the results from the NIMH study, we analyzed PANSS scores and time to discontinuation separately. Because all patients in the CATIE sample were undergoing treatment and the time and number of PANSS evaluations in the study varied considerably between participants, it was not possible to utilize the same analytic approach as we used for the placebo-controlled NIMH study. We therefore used change in PANSS scores (between the first and last available evaluations) for each participant and employed a general linear model to examine the effect of KCNH2 genotype while controlling for covariates such as age, sex, drug clearance, medications, and type of drug. As we only needed to fit a main-effect model for genotype on change in PANSS ratings in the CATIE data, there was a need for a reference genotype comparison group (here, it was TT genotypes). As a secondary analysis aimed at more closely reproducing NIMH inpatient trial analyses, we treated the first available PANSS rating as “before treatment” and the last rating as “after treatment” to test for a genotype-by-treatment interaction using a general linear model. We performed this analysis for all participants with clearance data (N=364) and separately only on those participants who completed the phase 1/1A trial (N=157). Because most patients who did not complete the CATIE study discontinued treatment for reasons other than efficacy, the specific analysis of change in PANSS ratings in patients who completed the trial (i.e., patients who continued the same treatment until the end of phase 1/1A) is likely to be more analogous to the treatment-effect analysis of patients in the NIMH placebo-controlled trial. Because the mean and variation of drug clearance varied among drugs, we assigned an ordinal measure of 1 to 4 according to the quartile distribution of each drug clearance to make the measurement of clearance comparable between drugs.
We also examined time to discontinuation of medication, which was the primary outcome measure in the CATIE study. Time to discontinuation, however, is a problematic measure in a pharmacogenetic analysis of therapeutic response because it is confounded by many factors, including side effects, patient preference, and other subjective individual considerations. Patients in the CATIE study were allowed to stop medication for any reason at any time. An illustration of the uncertainties inherent in time to discontinuation as an efficacy measure is that it was not predicted by drug clearance rate, which did predict symptom change during treatment (
20). In addition, the reasons for discontinuation and time to discontinuation for any reason in each drug arm differed substantially. Nevertheless, we assessed genotype effect on time to discontinuation in the olanzapine response data. Olanzapine had the longest time to discontinuation in the study and the least number of discontinuations for reasons other than efficacy; thus, time to discontinuation of olanzapine may better reflect variable efficacy. Furthermore, we had previously curated the olanzapine data to exclude nonadherent participants and participants receiving other medications as part of the clearance determination (
20). The Cox proportional hazards model was used to analyze the time to discontinuation data while controlling for age, sex, duration of illness, and drug clearance.
Results
The demographic characteristics of the NIMH sample are summarized in
Table 1 by rs1036145 genotype, a SNP showing effects on treatment in this sample. Age at admission (p<0.032) and duration of illness (p<0.001) were significantly different between genotype groups. The genotype groups also differed slightly in gender proportions and in proportion of smokers. These effects are unlikely to account for the genotype effects on outcome variables and were used as covariates in the final analyses. In particular, the longer duration of illness of a genotype group would not be expected to predict better response. No significant differences by genotype groups were observed for IQ, years of education, and prestudy antipsychotic dosage in chlorpromazine equivalents (
Table 1).
The demographic characteristics of the CATIE sample genotype are summarized in
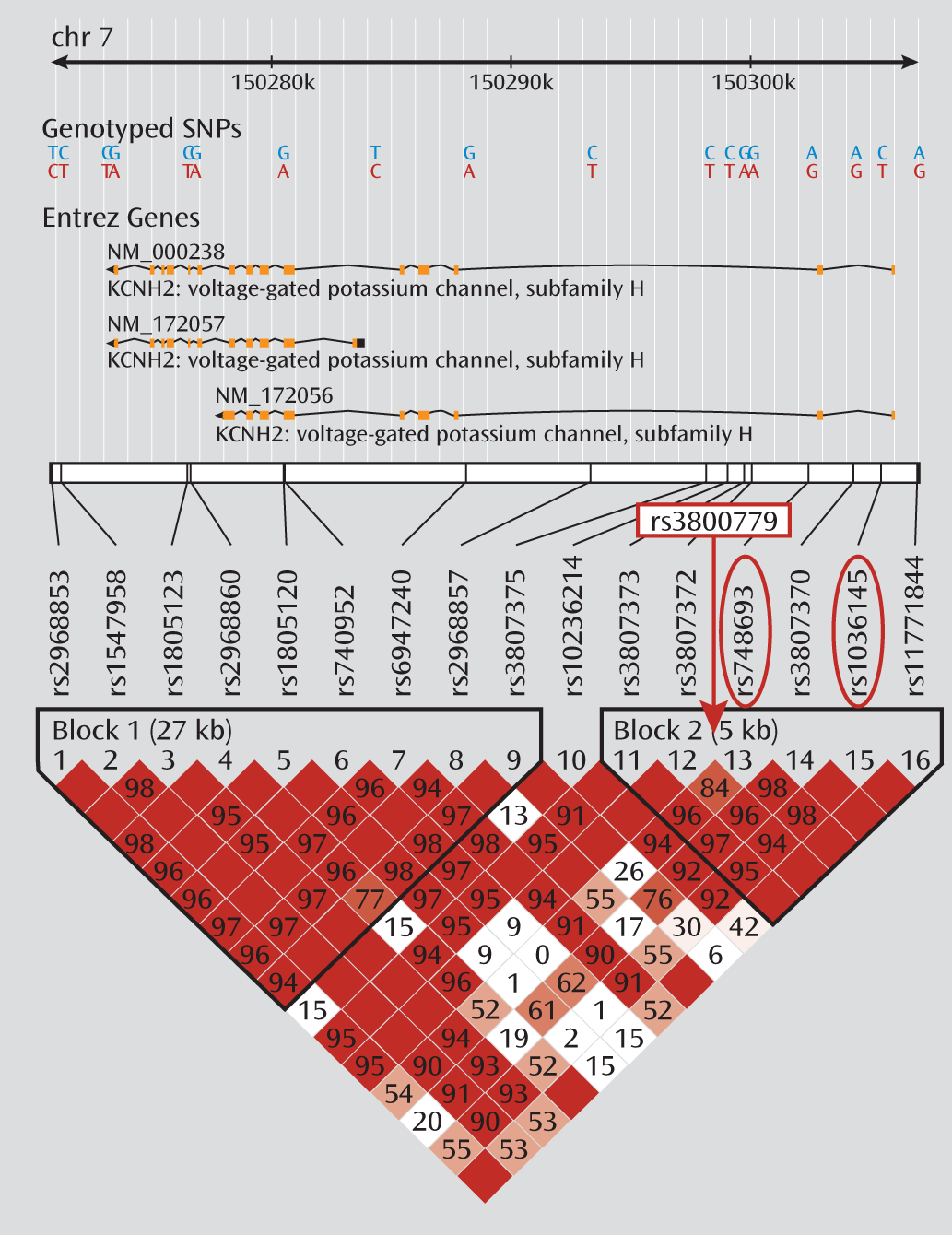
Table 2. No significant differences between groups were observed for mean dosage, years of education, age at illness onset, or duration of illness. Smoking status also did not differ by genotype. The three genotyped SNPs were in moderate to strong linkage disequilibrium in both samples (r
2 values between 0.7 and 0.9) (
Figure 1). Because of the especially strong linkage disequilibrium between rs748693 and rs1036145 (r
2=0.9), we present the data only for rs3800779 and rs1036145, as the results for rs748693 are similar.
In the NIMH study, a significant effect of antipsychotic treatment on PANSS scores was observed in various scales and clusters of the PANSS (
Table 3). Overall, medications had a significant impact on the alleviation of positive syndrome ratings (β=–1.357, p=0.0007) and general psychopathology (β=–2.285, p=0.0093); an effect was also observed on the decrease in negative syndrome (β=–1.084, p=0.0124), but it did not survive correction for multiple testing. Analysis of the PANSS clusters also revealed significant changes in thought disturbance (β=–0.893, p=0.0004) and activation (β=–0.684, p=0.0018).
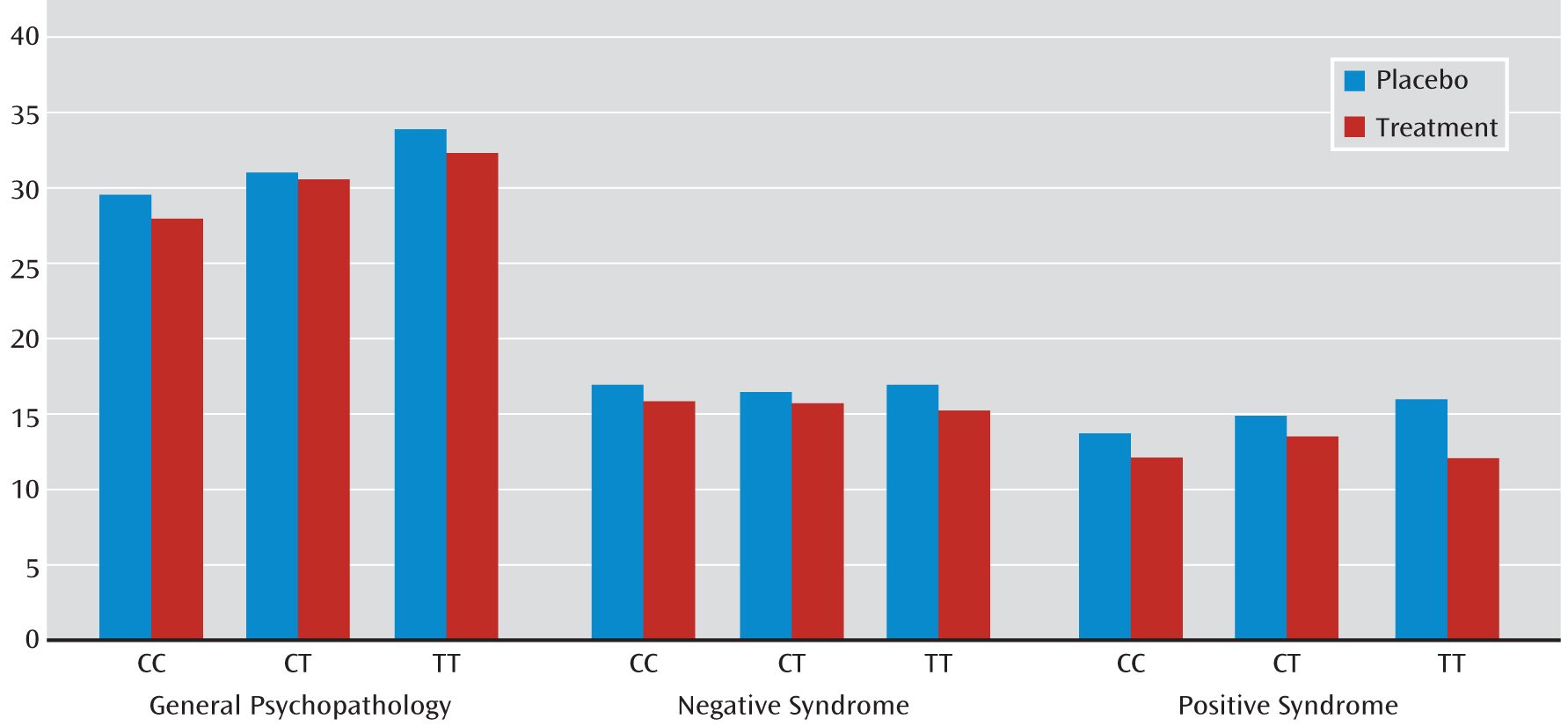
We observed a significant interaction between rs1036145 genotype and antipsychotic treatment on the improvement in PANSS ratings of positive syndrome (p=0.0395) and a nonsignificant interaction on general psychopathology (p=0.1516) in the NIMH cohort (
Table 4). In general, individuals with the TT genotype had a better response to antipsychotic treatment. Specifically, individuals who were homozygous for the T allele (i.e., genotype TT), the allele associated with increased schizophrenia risk and isoform 3.1 expression in the brain, had a significant reduction in ratings of positive syndrome (β=–2.69, p=0.0005) and general psychopathology (β=–5.41, p=0.0065) and a nonsignificant effect on ratings of negative syndrome (β=–1.87, p=0.0446) when they were taking medications (
Table 5 and
Figure 2). The magnitude of genotype effect on the change in each of these syndrome scores was greater than the effect of medication. We did not observe a significant interaction between rs3800779 genotype and antipsychotic treatment on any of three PANSS ratings (p>0.2), but again, TT genotypes of rs3800779 showed stronger effects on reduction in the positive syndrome and in general psychopathology than other genotype groups, despite being the smallest sample in the comparison.
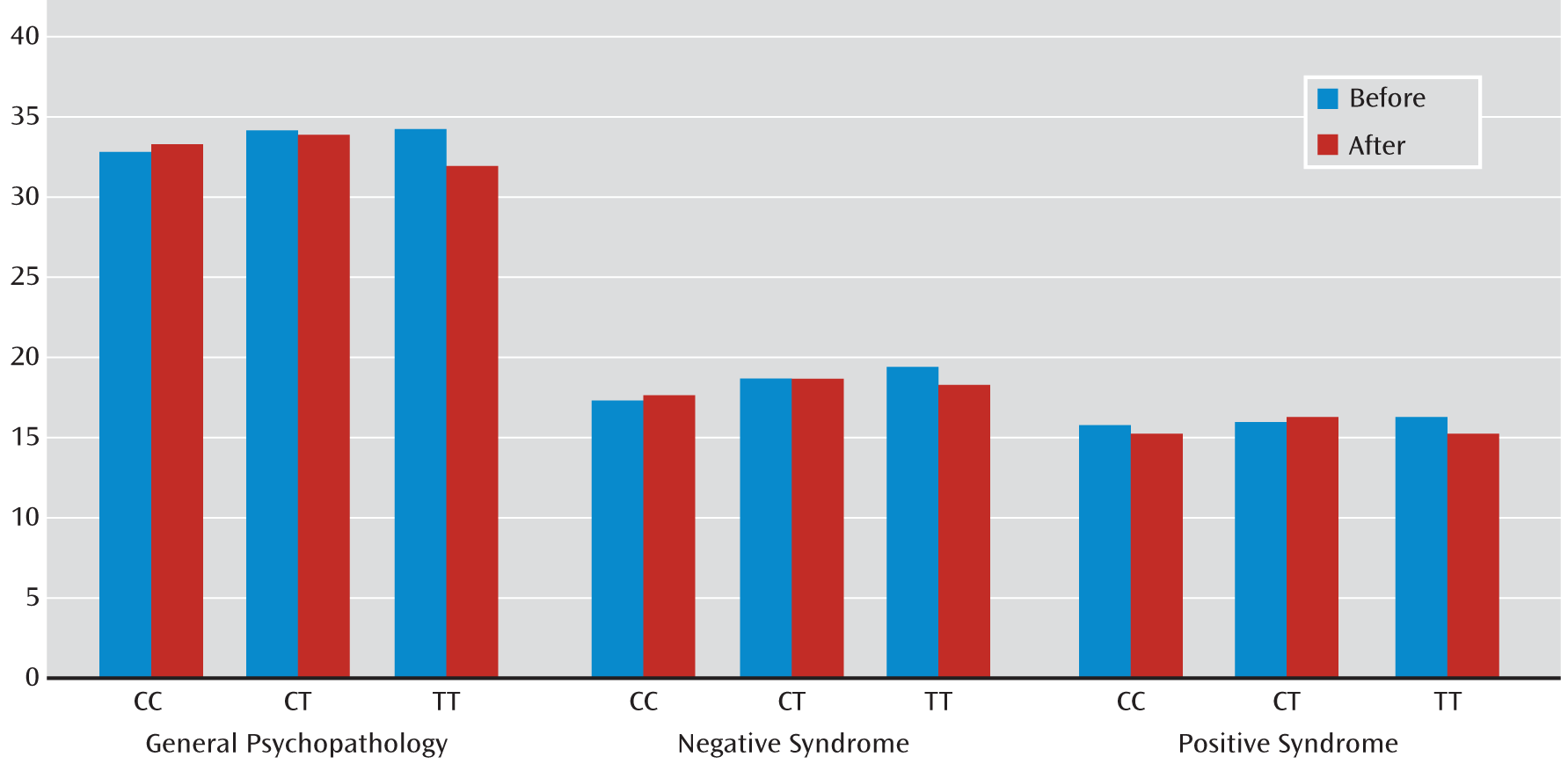
Similar genotypic effects on the improvement of PANSS scores were also observed in the larger CATIE cohort for both SNPs (
Table 6). The change in PANSS positive syndrome score varied significantly with the rs3800779 (p=0.0097) and rs1036145 (p=0.0025) genotypes. Compared with homozygotes for the risk allele, there were fewer changes in PANSS positive syndrome ratings for genotype GT at rs3800779 (β=–1.285, p=0.087) and for CT at rs1036145 (β=–1.306, p=0.0825) (
Table 6). The change in PANSS general psychopathology score was also associated with the rs3800779 (p=0.0425) and rs1036145 (p=0.066) genotypes. Compared with homozygotes for the risk allele, there were fewer changes in ratings of PANSS general psychopathology for genotype GT at rs3800779 (β=–2.48, p=0.0382) and for CT at rs1036145 (β=–2.499, p=0.0402) (
Table 6). (A summary of PANSS ratings is provided in
Table 5 and
Figure 3.) However, the change in negative syndrome with either SNP in the CATIE study did not reach significance.
In the secondary analysis of the CATIE PANSS ratings, modeled on the NIMH analysis, similar associations of genotype effect on treatment-associated change in PANSS ratings of positive syndrome and general psychopathology were noted at both rs3800779 and rs1036145, and limited genotype-by-treatment interactions were found in the subsample that completed the study (
Table 7). Again, in general, individuals with risk genotypes appeared to have a better response to antipsychotic treatment. In the subgroup analysis of individuals who completed the phase 1/1A trial, rs1036145 genotype-by-treatment interactions were observed for PANSS ratings of both positive syndrome (p=0.0756) and general psychopathology (p=0.0106). Individuals with the TT risk genotype had highly significant reductions in ratings of positive syndrome (β=–3.52, p=0.0004) and general psychopathology (β=–6.87, p=9.2W10
–6) that were not seen in the other genotype groups.
Analysis of the time to discontinuation of olanzapine using clearance data from the CATIE sample (N=89) also indicated that
KCNH2 genotype affected this primary outcome measure of response to antipsychotic treatment in this study (
Table 8). Overall time to discontinuation of medication at rs1036145 varied by genotype, but this relationship fell short of significance (p=0.0633). Compared with nonrisk allele homozygous individuals, the small group of individuals (N=10) with TT genotypes at rs1036145 were nearly about one-fifth as likely to have discontinued their medication (hazard ratio=0.208, p=0.033).
Discussion
This study provides consistent evidence of an association between variations in KCNH2 and short- and long-term clinical response to antipsychotic drugs in patients with schizophrenia. In two different treatment cohorts that were ascertained and examined with notably differing approaches, KCNH2 genotypes previously associated with schizophrenia and with expression of a brain-selective isoform of this potassium channel modulated the therapeutic effects of antipsychotic drugs.
KCNH2 genotype (rs1036145) had a significant impact on the response to antipsychotic treatment in the NIMH cohort in two separate groups: those receiving placebo first and those receiving active drug first. Patients who were homozygous for the risk-associated allele (the TT genotype) showed a greater change in ratings of positive syndrome and general psychopathology relative to carriers of the C allele. This effect of genotype, which remained significant after correcting for multiple testing and adjusting for covariates of age, duration of illness, sex, and IQ, is noteworthy considering the relatively small sample size of this cohort and the variation in the protocol sequence of the two groups within this study. In fact, the effect of genotype on treatment-associated symptom reduction was larger than the effect of treatment.
The pharmacogenetic associations and their directionality were confirmed and further extended in the analysis of the larger CATIE cohort. Interestingly, the significant changes in positive syndrome and general psychopathology were observed in the homozygous risk-associated alleles at both SNPs, rs3800779-TT and rs1036145-TT, the latter showing the stronger statistical significance, in line with the results obtained in the smaller NIMH trial. The impact of this finding in two independent cohorts is underscored by the fact that these trials were performed with two different clinical and experimental models and different outcome measures—one an outpatient parallel-cohort drug-only design and the other a double-blind, placebo-controlled inpatient crossover study. The effect size of the association is also noteworthy. In the placebo-controlled NIMH study, individuals homozygous for the risk allele showed almost twice the effect size of response change compared with the other genotype groups. In the CATIE study, individuals with the risk genotypes showed one-fourth to one-fifth the likelihood of discontinuing olanzapine, a primary outcome measure of the trial, although the magnitude of this effect is qualified by the relatively small homozygous genotype sample (N=10).
In both the NIMH and CATIE samples, individuals with risk-associated genotypes, which predict increased expression of isoform 3.1 of KCNH2, tended to have a better response to antipsychotic drug therapy. In other words, the greater the putative expression of the novel schizophrenia-associated isoform, the larger the therapeutic effect was. Interestingly, the effect is less obvious in heterozygous individuals, perhaps suggesting a recessive genetic mechanism on antipsychotic drug response. In earlier work, the effect of genotype on the simpler molecular phenotype of gene expression appeared to be additive. We believe it is not surprising that the penetrance of this molecular effect is not as strongly predictive at the clinical symptom level, and homozygosity may be a more realistic predictor of a consistent effect.
Regardless of the genetic model, it is tempting to conclude that our pharmacogenetic results reflect the ability of antipsychotics to bind to this novel
KCNH2 potassium channel and that their therapeutic effects are at least in part related to this effect (
8,
9). This conclusion, however, must be viewed with caution. Clearly, many drugs bind to hERG channels, and only a few of them are antipsychotics. hERG channels are heterodimers of various hERG components, and the combinations of full-length and 3.1 isoform channels may be critical for understanding the action in the brain of varying effects of hERG-active drugs. Moreover, if the antipsychotic effect of drugs is selectively related to their modulation of channels containing 3.1 isoforms, then drugs that affect only 3.1-lacking hERG channels might not be antipsychotic. The physiological effect of the 3.1 isoform in neuronal culture (
15) and also in an inducible
KCNH2 3.1 transgenic mouse we created (
23) suggests that the optimum therapeutic effect would not be channel blockade but restoration of normal hERG1 potassium conductance kinetics (
15). Further research on the mechanisms through which antipsychotics modulate the 3.1 isoform-related potassium channel may provide more reliable information on whether antipsychotics act in part by this mechanism.
An alternative indirect mechanism for the relationship between
KCNH2 genotype and antipsychotic drug response might also be considered. Since hERG1 channels are expressed on dopamine neurons (
13), it is conceivable that their relationship to antipsychotic drug action is by means of an indirect mechanism related to the firing characteristics of these neurons. If dopamine neurons are overactive in psychotic patients, as is implicated by results of in vivo imaging studies (
24,
25), increased
KCNH2 3.1 expression may, at least in part, drive this overactivity, which is compensated by postsynaptic blockade of D
2 receptors. By either mechanism, our results raise intriguing questions for further work and for the potential of
KCNH2 3.1 selective drugs to be novel antipsychotic therapies and encourage the pursuit of
KCNH2 3.1 as a potential target for the development of new therapeutic agents.
Each of the clinical trials in this report has limitations. The NIMH study involved a relatively small number of patients. However, it employed a double-blind, placebo-controlled crossover design in a stable inpatient population with chronic and unremitting schizophrenia in which medication intake was strictly monitored by experienced nursing staff in a highly structured research environment. A limitation of the CATIE cohort is the relatively uncontrolled outpatient nature of the study, which had no stringent controls for medication adherence. This problem was addressed by determination of antipsychotic medication levels, and only those individuals in whom drug levels were sufficient to determine plasma clearance rates were included in this analysis.
One of the main concerns related to the pharmacological action of antipsychotic drugs and to patient adherence has been the inconsistent therapeutic response observed between individuals (
26). While there are likely many factors relating to this variable treatment response, our data suggest that
KCNH2 genotype is one potentially important factor. Moreover, our findings support the hypothesis that the hERG1 activity of antipsychotics may be connected not only with their potential cardiovascular side effects (
27) but also with their therapeutic actions involving the brain-selective 3.1 isoform of
KCNH2 (
15,
16).
Acknowledgments
The authors thank the nursing staff on the 7-SE unit at the NIH Clinical Research Center for providing Positive and Negative Syndrome Scale ratings and Patrick Sullivan for performing the KCNH2 genotyping on the CATIE DNA samples.