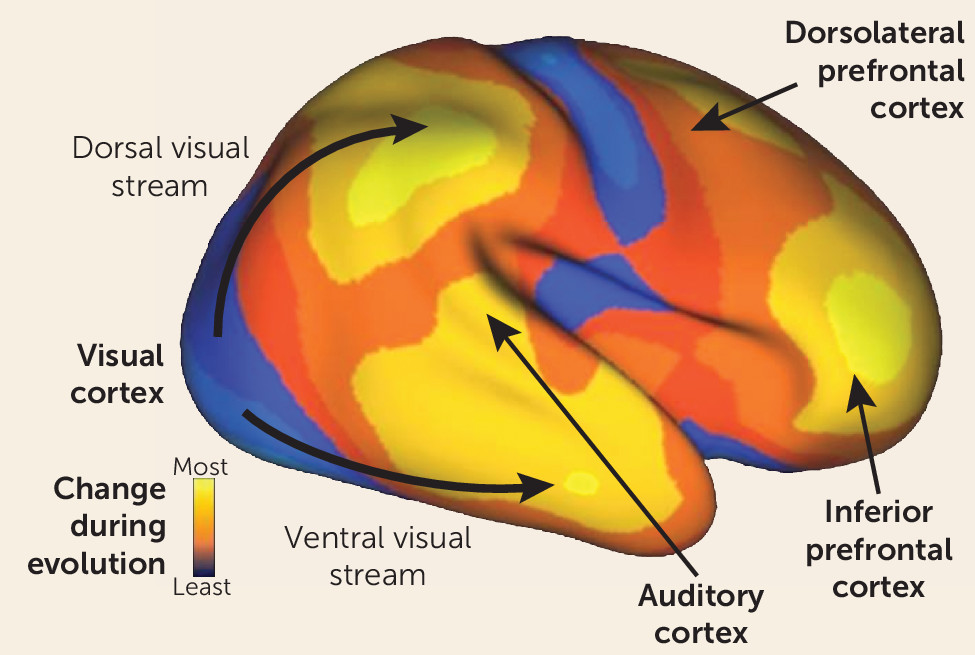
It is well known that humans are distinguished from other primates by our superior reasoning and problem-solving ability, both of which are related to a dramatic expansion in frontal brain regions as compared with other primates (
Figure 1). It is less appreciated that an equally marked evolutionary expansion has occurred in the human auditory system (
5). This expansion underlies not only our unique linguistic abilities but also our greatly expanded ability to appreciate pitch, rhythm, and other musical features relative to other primates. Like other recently evolving regions, the auditory cortex completes its maturation late in human development (
5). Thus, like prefrontal regions, these systems may remain vulnerable to disruption even during late adolescence and early adulthood, when schizophrenia typically develops. In general, sensory systems play a dual role—first, to orient attention to critical regions and features of the environment, and second, to decode the information emanating from those regions to enable subsequent voluntary processing. In schizophrenia, both the attentional and informational roles of the sensory systems are impaired, contributing to the overall pattern of symptoms and neurocognitive deficits we frequently associate with the disorder.
Sensory and Sensorimotor Gating Dysfunction
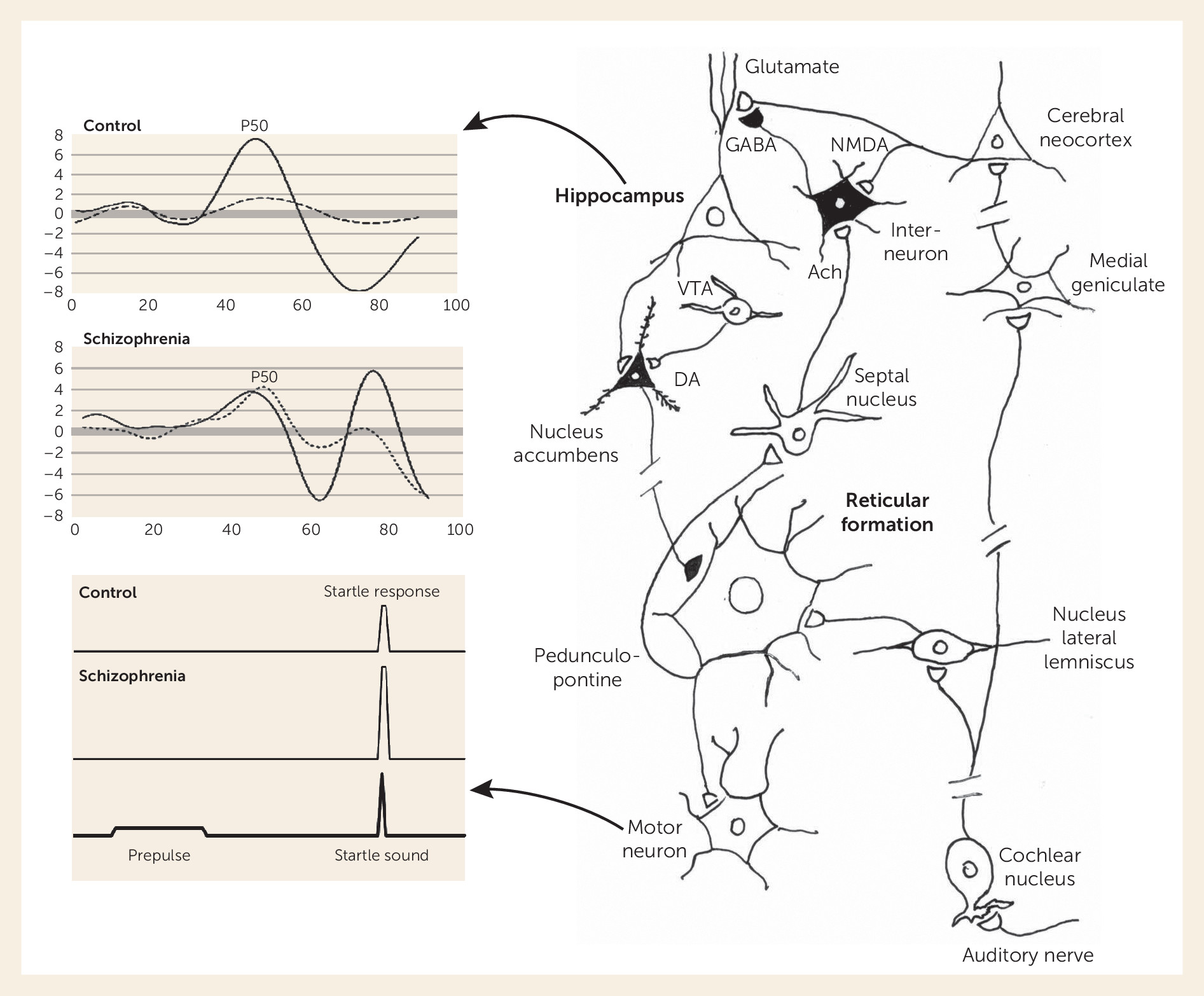
Auditory gating relies on the interplay of auditory input and brainstem reticular formation function. Reticular neurons have rapidly habituating responses to repeated stimulation and thus form the brain’s initial sensory gating mechanism, which prevents other brain regions from being flooded with repeated and presumed less important sensory information, such as in a noisy environment. The habituation to repeated stimulation leaves the brain ready to respond to more novel and thus potentially more meaningful sensory stimuli.
The effect of the brainstem’s habituation is observed experimentally in two ways. First, a descending spinal pathway from the larger reticular neurons initiates the startle response. A weak tone initiates habituation of the reticular neurons’ response to a subsequent louder tone, leading to the phenomenon of prepulse inhibition (PPI) of the acoustic startle response (
7,
8).
In parallel, projections from other reticular neurons ascend to the midbrain, where they contact cholinergic neurons in the medial septal nucleus, which in turn project to the hippocampus, thalamus, and neocortex. These cholinergic projections activate inhibitory interneurons in the hippocampus and other areas that modulate local excitatory glutamatergic neurotransmission (
9). The hippocampus then inhibits response to less important information coming from the auditory cortex and other sensory regions, funneled to it through the superior temporal gyrus. In humans, the largest initial cerebral response to an auditory stimulus is a positive wave that occurs at 50 ms and is thus termed the P50 potential. The P50 response to the second of paired stimuli is decreased relative to the first in healthy persons, an example of inhibitory sensory gating (
10).
Deficits in the integrity of both PPI and P50 gating in schizophrenia have been extensively replicated since they were first demonstrated, pointing to dysfunction even within these low-level circuits. The deficit in PPI is most prominent in schizophrenia when the weaker tone precedes the startling sound by 60 ms (
7). The deficit is more pronounced in women, who normally show less inhibition than men. Patients with schizophrenia, either unmedicated or treated with most antipsychotic drugs, also have reduced inhibition of the P50 auditory evoked potential (see
Figure 2). The inhibition of the second stimulus is maximal at 500 ms, later than the maximal PPI interval of 60 ms. In many patients, the two deficits do not occur together, and thus they represent distinct aspects of pathophysiology (
11). The N100 also shows impaired gating in schizophrenia (
12).
For both PPI and P50 gating, despite the origin of the effect in reticular brainstem neurons, the response engages an entire network of brain circuits—the dopaminergic circuits of the basal ganglia for PPI and the basal cholinergic nuclei and cortex for P50 gating (
8,
9). Rodent models of P50 gating emphasize the response in the hippocampus, but sources for human P50 have been found in the superior temporal gyrus, and cortical rhythms are altered during the sensory gating paradigm, with a decrease in EEG beta waves (
13). Deficits in the response to repeated sounds have also been localized to the hippocampus and dorsal lateral prefrontal cortex by both dipole analysis and functional MRI (fMRI) (
14,
15).
P50 inhibitory gating deficits in schizophrenia have been associated with the frequency and intensity of auditory hallucinations and with neurocognitive measures of decreased vigilance and attention, consistent with the role of inhibitory sensory gating in protecting the brain from being responsive to extraneous stimuli while it is trying to focus attention on a task (
16,
17). The association between patients’ self-report of sensory phenomena and physiological measures is confounded by the patients’ psychopathology (
18). Thus, several investigations in patients have not shown association between P50 inhibition and self-reports of sensory gating difficulties (
19), but in schizotypal adults, deficiencies in P50 inhibition are associated with difficulties in “hearing everything at once” and with a sense of unreality, including perceptual anomalies and magical beliefs (
20,
21). PPI deficits have been correlated with deficient social perception (
22).
Genetic Endophenotypes
Inhibitory gating measures have also contributed significantly to the search for genes involved in schizophrenia. Deficits in both PPI and P50 gating are found in schizotypal individuals who have a family history of schizophrenia and in adolescents at high risk for schizophrenia (
23–
25). As a complex genetic illness, schizophrenia has not been linked to a single major gene. However, these specific endophenotype measures, each thought to underlie one aspect of the genetic basis of schizophrenia, were hypothesized to have a closer relationship to particular elements of genetic risk (
20).
In families affected with multiple cases of schizophrenia, the deficit in P50 gating segregates as an autosomal dominant trait and is linked to the CHRNA7 gene complex, which forms the α7-nicotinic acetylcholine receptor (
26). Polymorphisms of CHRNA7 have been associated with PPI as well (
27). Another related gene is NRG1, which forms neuregulin (
28,
29). Neuregulin helps assemble both α-nicotinic and
N-methyl-
d-aspartate-type glutamate receptors (NMDARs) (
30), and thus its association with both sensory processing deficits is not surprising. The α7-nicotinic receptor is found presynaptically on glutamate nerve terminals and postsynaptically on inhibitory interneurons (see
Figure 2), particularly in the hippocampus and nucleus reticularis thalami (
9). Thus, there is convergence between the genetic findings and the neurobiological model, corroborated by postmortem findings of decreased α7-nicotinic receptors in schizophrenia (
31). As the scope of genomic analysis has increased, both deficits have been associated with entire networks of genetic variants in schizophrenia in the NIMH Consortium on the Genetics of Schizophrenia study, which examines the relationships of these phenotypes to the cognitive and genetic aspects of schizophrenia (
28).
Drug Development
Although schizophrenia cannot be reproduced in animals, mouse and rat analogues of PPI and P50 deficits can be produced. Increased dopaminergic neurotransmission from stimulants and psychotomimetic antagonists of NMDARs such as phencyclidine (PCP) induce the deficit pharmacologically (
8). Deficits in CHRNA7 null mutant mice for P50 gating (
32) and in NRG1 null mutants for PPI have also been reported (
8). Intriguingly, the NRG1-induced deficits do not appear until adolescence (
33), modeling the time course for onset of schizophrenia (
34).
Animal models were first used to examine effects of nicotine and clozapine, with good corroboration in clinical observations (
10). Cigarette smoking increases P50 inhibition and PPI in schizophrenia (
35). Further animal and clinical investigation confirmed nicotine's activation of α7-nicotinic receptors as the mechanism of its effects on P50 inhibition (
36,
37). Clozapine, unlike other antipsychotics, significantly normalizes P50 inhibition, through increased release of acetylcholine in the hippocampus and the resultant indirect activation of α7-nicotinic receptors on inhibitory interneurons (
38).
Investigational therapeutics followed, to develop specific α7-nicotinic receptor agonists to enhance cognition. The initial proof of principle for several of the agents included the demonstration of normalization of P50 inhibition in schizophrenia (
39). Results of human studies have been mixed, with some but not all showing enhancement of cognition and relief of negative symptoms (
39–
41). Nevertheless, at least one compound is currently in late-stage clinical testing, with definitive results expected within the next several years (
42).
Tone Matching Ability
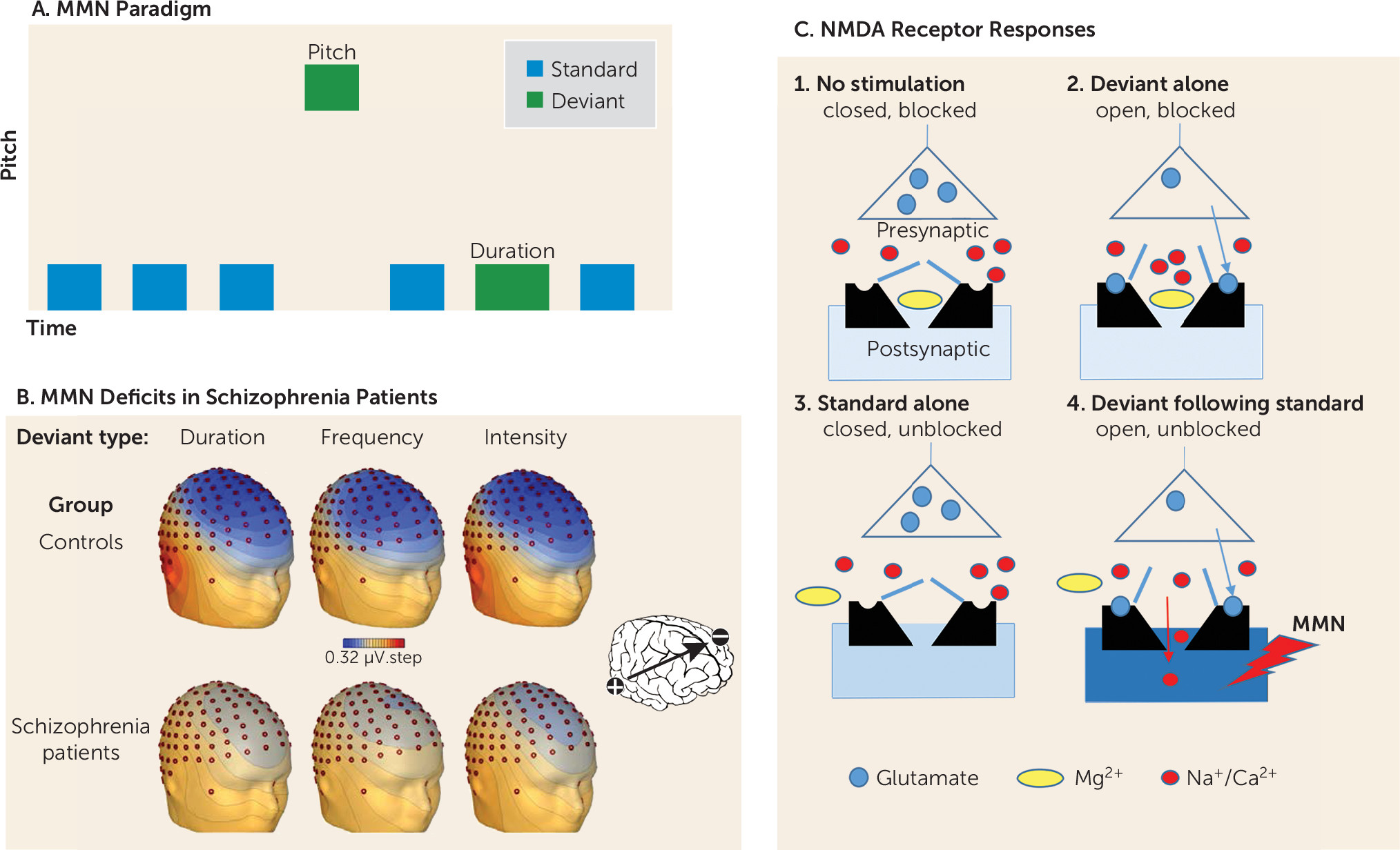
One of the most widely studied measures of cortical auditory dysfunction is the mismatch negativity (MMN), which is elicited whenever a stimulus differs from what is expected based on recent auditory experience. MMN is elicited most frequently in an auditory “oddball” task, in which a series of repetitive stimuli is interrupted infrequently by a physically deviant “oddball” stimulus. Deviant stimuli can differ on any of several physical dimensions, including pitch, duration, intensity, and location (
53) (
Figure 3A).
MMN has been tied to impaired neurotransmission at NMDARs in both intracranial (
54) and surface (
55) recordings in nonhuman primates and in ketamine challenge studies in healthy human volunteers (
3). Unlike PPI, MMN generation is little affected by either dopaminergic or serotonergic manipulations (
56). However, as with P50 gating, ketamine-induced deficits in MMN generation are reversed by nicotinic agonists (
57), possibly by presynaptic stimulation of nicotinic receptors on the glutamate nerve terminals (
40,
58), with similar effects observed in schizophrenia (
39). Other compounds that may reverse MMN deficits in schizophrenia include
N-acetylcysteine, a precursor of the brain antioxidant glutathione (
59), and
d-serine, an endogenous NMDAR modulator (
60).
Deficits in MMN generation were first reported in the early 1990s, and this finding has since been a widely replicated neurophysiological finding in schizophrenia (
3,
61–
63) (
Figure 3B). Deficits in MMN generation correlate highly with overall level of function (
64) and age at which symptoms of the illness first appeared (
65). Like P50 and PPI deficits, MMN deficits are heritable across the general population (
66). In persons at high clinical risk for schizophrenia, MMN has emerged as a predictor of who will progress to schizophrenia and therefore may require the most intensive remediation (
67). Although the majority of studies of auditory dysfunction in schizophrenia have used neurophysiological measures such as MMN, similar deficits may be detected using fMRI to evaluate response to stimulus change within auditory cortical regions (
68).
In keeping with the continued maturation of the auditory cortex in adolescence, MMN impairments continue to develop during the initial stages of the illness (
69) in parallel with progressive volume reduction in auditory brain regions (
70). In addition to MMN deficits, patients with schizophrenia also show deficits in the generation of other cortical auditory responses, such as the auditory steady-state response to rapidly presented stimuli (
71). Patients fail to entrain their neuronal response to the rhythm of repeatedly presented stimuli, and so make less efficient use of sensory processing resources (
72).
On the behavioral level, deficits in MMN generation are associated with impairments of basic auditory discrimination, such as tone matching (
3) and auditory spatial discrimination (
49), both underappreciated aspects of the clinical features of schizophrenia. Important social information, including emotion and attitude, is conveyed by variation in vocal intonation (“prosody”). Consequently, impaired ability to detect vocal intonation may contribute significantly to impaired social function (
73).
Deficits in tone matching contribute significantly to the ability to detect emotional prosody (
74,
75), as well as to communications such as sarcasm that require the individual to appreciate what another person may be thinking (
76,
77), a trait called “theory of mind,” a form of empathy. On standardized tests of musical ability, such as the Montreal Battery for Evaluation of Amusia (
78), nearly 50% of schizophrenia patients show deficits, compared with only 10% of the general population (
79).
Deficits in MMN generation, particularly to duration deviants, are also prominent in developmental dyslexia, reflecting impaired ability to perform phonological operations (i.e., “sounding out” words) required for successful reading (
80). Recently, similar reductions in phonological reading ability have been observed in patients with established, but not prodromal, schizophrenia, which suggests that progressive reductions in auditory function during the peri-onset period may result in a significant regression in reading ability in schizophrenia from premorbid levels (
81). Such findings may identify pathways through which deficits in sensory function lead to impaired functional outcome when schizophrenia itself develops later on.
Although neural circuitry underlying MMN generation has not been fully determined, specific properties of the NMDAR appear to play a key role. NMDARs are unique in that they are controlled by both the resting membrane potential of the postsynaptic cell and glutamate release from the presynaptic cell. Specifically, at resting membrane potential (approximately −65 mV), NMDARs are blocked by Mg, which binds to a site within the NMDA channel (
Figure 3C). Thus, even if glutamate is released from presynaptic terminals in response to a stimulus, no current flows through NMDAR channels even though they are open.
A proposed model of MMN generation (
Figure 3C) is that repetitive standard stimuli induce subthreshold depolarization of the resting membrane potential in a population of neurons sensitive to stimulus properties that differ from those of the standard stimuli, leading to unblocking of the channels even though the channels are still closed. Once receptors are unblocked, presentation of the deviant stimulus leads to opening of the channel. Current flow through the open, unblocked NMDAR channel on neurons in the auditory cortex leads to the generation of the surface MMN potential. Although this phenomenon can be studied best in humans by use of sensory stimulation and auditory evoked potentials, similar phenomena occur elsewhere in the brain and lead to the types of cognitive disturbances observed in schizophrenia.