Major depressive disorder has a lifetime prevalence of 16.2%, causes greater total morbidity, loss of productivity, and suicide than any other noncommunicable disorder, and contributes significantly to decreased quality of life (
1,
2). Despite significant advances in neuroscience, treatment development has lagged, primarily because of a lack of applicable clinical neuroimaging or other biomarkers: no widely accepted biomarkers are available to assist diagnostics or treatment choice for individual patients. The timely selection of the optimal treatment for patients with depression is critical to improving remission rates. Owing to the biological heterogeneity and variable symptom presentation of depression, it is unlikely that a single clinical or biological marker can guide treatment selection. Rather, multiple biological measures may be needed to refine our understanding of the underlying pathology and provide more reliable markers to guide treatment. Unfortunately, predictor research has been limited by the use of a single clinical or biological marker and as a result has explained a small degree of variance.
As has been highlighted previously (
3), neuroimaging technologies have the potential to identify objective neurobiological markers reflecting underlying pathophysiologic processes in a given psychiatric illness, which can ultimately facilitate the development of personalized treatments based on a better understanding of these underlying processes. Moreover, with the advancement of different types of neuroimaging technologies and data analytic techniques, there are now enormous opportunities to adopt multimodal neuroimaging approaches to examine the functional and structural integrity of parallel distributed neural circuits implicated in a given illness. In turn, this approach can both help identify multiple biomarkers reflecting underlying pathophysiologic processes in illnesses such as depression, and help in determining the extent to which such biomarkers can serve as predictors of treatment response in the illness. Typically, however, studies in depressed individuals have focused on examination of one neural circuit of interest and have not employed multimodal neuroimaging techniques to refine our understanding and thereby provide more reliable biomarkers of functional and structural abnormalities in parallel neural circuits of interest. Furthermore, a relatively small number of studies have used neuroimaging to help identify biomarker predictors of antidepressant response in depression, and, as noted, they have focused primarily on one neural circuit. Moreover, no study to date has focused on identifying neuroimaging moderators, pretreatment variables that predict differential treatment response, or neuroimaging temporal mediators, variables whose change early during treatment is associated with future treatment outcome, in depression. Identification of the former can improve treatment selection for depressed individuals. Identification of the latter can help stop ineffective treatment early, in addition to facilitating our understanding of early, even if not longer-term, causal neural mechanisms of treatment response in these individuals (
4). Elucidating neuroimaging moderators and mediators can thus provide not only valuable insights into neural mechanisms of illness but also valuable clinical information to help guide the choice of treatment for these individuals early on.
In this review, we first examine the extent to which multimodal neuroimaging techniques can be used to identify biomarkers reflecting key underlying pathophysiologic processes in depression. We describe two parallel neural circuits, namely, implicit emotion regulation and reward neural circuits, that are relevant to understanding pathophysiologic processes underlying core symptom dimensions in depression. We examine the extent to which these neural circuits are modulated by different neurotransmitter systems, and the nature of functional, gray and white matter structural, and resting-state functional connectivity, as well as blood flow abnormalities, in these neural circuits in depressed individuals. We then examine the extent to which biomarkers reflecting functional and structural abnormalities in these circuits may have utility as predictors and, more importantly, as moderators and mediators of treatment response to specific antidepressant treatments. We end by discussing future directions for neuroimaging studies of treatment response prediction in depression.
Neural Circuits Implicated in the Pathophysiology of Depression
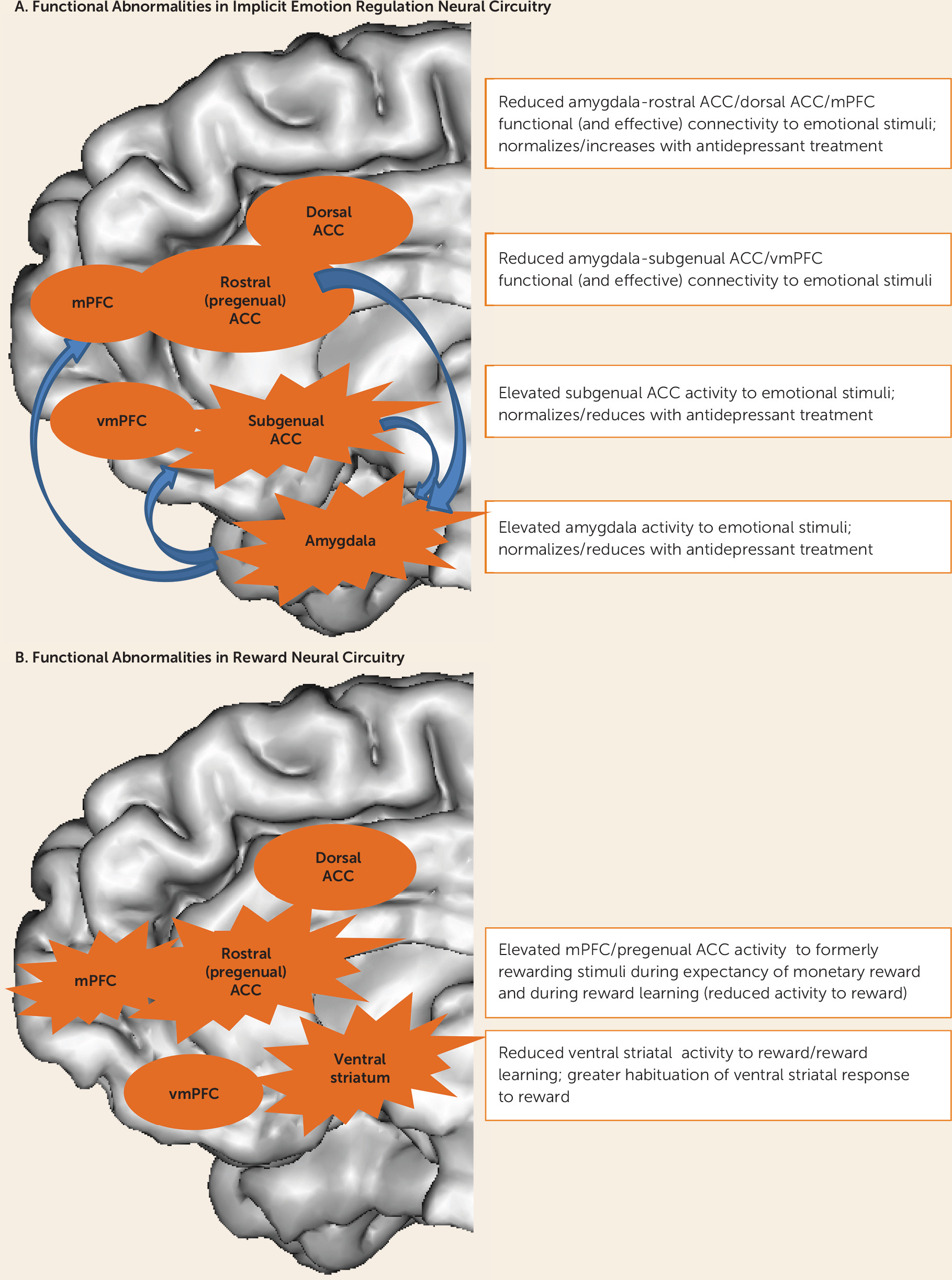
Core depressive symptom dimensions, including persistent low mood, anxiety, and anhedonia, reflect predominant features of dysfunctional emotion regulation and reward processing. While abnormalities in multiple distributed neural circuits underlying different levels of effortful and implicit emotion regulation and reward processing are probably implicated in the pathophysiology of depression and other affective disorders (
3,
5), the most consistent findings involve two patterns of distinct functional abnormalities: those in 1) serotonergically modulated implicit emotion regulation neural circuitry centered on the amygdala and different medial prefrontal cortical regions, and 2) dopaminergically modulated reward neural circuitry centered on the ventral striatum and medial prefrontal cortex (
6–
9) (
Figure 1). Abnormalities in these parallel neural circuits may be associated with different symptom dimensions and therefore guide appropriate treatment selection. For example, abnormalities in implicit emotion regulation circuitry may be associated with persistent low mood and anxiety, while abnormalities in reward neural circuitry may result in apathy and anhedonia (
10,
11). Examining relationships between abnormalities in these neural circuits and different symptom dimensions in depressed individuals also parallels the dimensional approach of Research Domain Criteria advocated by the National Institute of Mental Health (
12) and can ultimately identify critical brain-behavior relationships that may transcend conventional diagnostic categories of psychiatric illness. For example, abnormalities in implicit emotion regulation circuitry may be associated with behaviors linked to constructs in the negative valence systems domain, such as acute and sustained threat, fear, and anxiety. On the other hand, abnormalities in reward circuitry may be associated with behaviors linked to constructs in the positive valence systems domain, such as reward expectancy and anhedonia. In the following sections, we describe in more detail the nature of these distributed neural circuits, their modulation by specific neurotransmitter systems, and the abnormalities in these circuits that are reported in depressed individuals.
Functional and Gray Matter Structural Abnormalities in Implicit Emotion Regulation and Reward Circuits in Depression
Some of the most consistent findings regarding functional abnormalities in implicit emotion regulation circuitry in depressed individuals are abnormally elevated activity in the amygdala and/or anterior cingulate cortex; reduced functional connectivity between the amygdala and medial prefrontal cortical regions in response to negative emotional stimuli; and, to a lesser extent, reduced activity in response to positive emotional stimuli (
83–
96). Interestingly, there is some evidence that abnormally reduced amygdala activity to positive emotional stimuli may be associated with anhedonia in depressed individuals (
97). There are inconsistent findings of either maintenance of abnormally elevated or abnormally reduced activity in this circuit, especially in the amygdala, during remission from depression (
98–
100). Longitudinal neuroimaging studies, however, have reported a normalization of abnormally elevated activity in this circuit in response to pharmacotherapy, especially treatment with SRIs (
24,
86–
88,
94,
96,
101,
102).
An increasing number of studies have reported functional abnormalities in reward circuitry in depressed adults. Depressed adults have been reported to show abnormally elevated rostral anterior cingulate cortical activity to previously rewarding stimuli (
103). Other studies of depression have reported either elevated (
104) or reduced (
105) activity in the pregenual and dorsal anterior cingulate cortices during expectancy of monetary reward, and a failure to deactivate the pregenual anterior cingulate cortex during reward learning (
7). Furthermore, elevated ventral/pregenual anterior cingulate cortical activity, together with reduced capacity to maintain ventral striatal activity to rewarding/positive emotional stimuli, has been reported to be associated with greater anhedonia in depressed adults (
106,
107). Some studies have indicated significantly reduced ventral striatal activity to rewarding stimuli and during reward learning in depressed compared with healthy adults (
7,
10,
43,
108,
109), and increased habituation of ventral striatal activity to reward (
110). Others have not found these associations (
104). Additional evidence suggests associations between greater anhedonia and diminished reward learning in depressed individuals (
111) and a normalization of functional abnormalities in reward circuitry with successful response to psychotherapy (
112) (
Figure 1).
While it is beyond the scope of this review to describe findings in detail, an extensive literature has documented abnormally reduced gray matter volume in regions overlapping with implicit emotion regulation and reward circuits in depressed individuals, in particular in the ventromedial prefrontal and anterior cingulate cortices and in subcortical regions (
113–
115). Studies examining cortical thickness, an index of neuronal integrity and arborization (
116), have reported 28% lower right cortical thickness in individuals at high risk for depression (
117).
Parallel Findings From Resting-State Functional Connectivity, Arterial Spin Labeling, and Diffusion Imaging Studies
There is a rapidly growing literature focusing on resting-state connectivity in a variety of neural regions and networks in depressed individuals. While there have been many inconsistent findings, key findings in neural circuits supporting implicit emotion regulation and reward processing indicate either abnormally increased or abnormally decreased resting-state connectivity between different anterior cingulate cortical subregions and other prefrontal cortical regions (
118,
119); abnormally reduced resting-state connectivity between subcortical regions, including between the amygdala and the striatum, and between the anterior cingulate and ventromedial prefrontal cortices (
86,
118–
122); decreased resting-state connectivity between the subgenual anterior cingulate cortex and cortical areas (
123); and abnormal patterns of resting-state connectivity between striatal and ventral prefrontal cortical regions and the whole brain (
124). Resting-state connectivity has also been reported to be abnormally increased across three large-scale networks, including the affective (subgenual) network, in depressed individuals (
125). Subcortical-anterior cingulate cortical resting-state connectivity has been shown to increase after SRI treatment (
86), although SRIs and antidepressant medications targeting catecholamine systems have been shown to decrease resting-state connectivity in healthy volunteers (
126).
A small number of studies have employed arterial spin labeling to examine regional cerebral blood flow in implicit emotion regulation neural circuitry in depression. A study comparing six patients with chronic treatment-resistant depression and six healthy subjects (
127) showed significantly greater resting cerebral blood flow in predominantly left-sided medial prefrontal cortical and subcortical regions in the depressed group. Another study (
128) reported that depressed individuals who responded to partial sleep deprivation had greater baseline amygdala blood flow relative to individuals who did not respond, and that cerebral blood flow in this region was reduced after treatment. In parallel, a study in healthy individuals (
129) showed that a single oral dose of the SRI citalopram was associated with reductions in cerebral blood flow in implicit emotion regulation circuitry regions, including the amygdala and the ventromedial prefrontal cortex. Another study (
130) found decreased perfusion in the prefrontal and anterior cingulate cortices in depressed adult nonremitters after a 6-month follow-up compared with healthy adults, but did not find any perfusion differences between depressed and healthy adults at baseline.
A meta-analysis of diffusion imaging data in mood disorders reported that 21 of 27 studies found significantly lower fractional anisotropy in the left and right frontal and temporal lobes or in white matter tracts connecting prefrontal cortical, subcortical, and other cortical regions in individuals with mood disorders relative to healthy volunteers (
131). More recent studies confirm this general pattern in individuals with, and those at risk for, depression (
132–
146), although there are some exceptions (
147).
Elucidating Abnormalities in Implicit Emotion Regulation and Reward Circuits in Depression: A Comparison with Bipolar Disorder
Specific themes emerge from the studies described above. These include, in implicit emotion regulation circuitry, abnormally elevated amygdala activity and reduced amygdala-medial prefrontal cortical functional connectivity to negative emotional stimuli in particular, paralleled by reductions in gray matter volumes in subcortical and prefrontal cortical regions. Resting-state functional connectivity studies indicate abnormally reduced, but also abnormally increased, resting-state functional connectivity between these regions, while arterial spin labeling studies report patterns of predominantly abnormally increased resting blood flow in the amygdala and in medial prefrontal cortical regions. Diffusion imaging findings indicate abnormally reduced fractional anisotropy in white matter tracts connecting these regions. These findings suggest compromised functioning in this circuitry, including insufficient regulation of subcortical structures such as the amygdala by medial prefrontal cortical regions, especially to negative emotional stimuli. The smaller number of findings in reward circuitry indicate abnormally elevated activity in anterior cingulate cortical subregions, especially the pregenual anterior cingulate cortex, during reward anticipation and receipt, and abnormal, predominantly reduced, ventral striatal activity during different stages of reward learning, although there are inconsistent findings.
Further understanding of these findings can be facilitated by comparing the functional and structural abnormalities in these circuits in depressed individuals with those observed in individuals with other mood disorders, in particular bipolar disorder. For example, findings suggest distinguishable functioning and structure in implicit emotion regulation circuitry in depressed individuals with major depressive disorder compared with depressed individuals with bipolar disorder; studies have also reported differential patterns of functional and white matter structural abnormalities in this circuitry in the two disorders (
85,
148,
149; see reference
150 for a review). These studies indicate greater amygdala activity in response to negative than to positive emotional stimuli, predominantly left-sided reductions in fractional anisotropy, and abnormally increased left-sided ventromedial prefrontal cortical-amygdala inverse functional connectivity to positive emotional stimuli in depressed individuals with major depressive disorder. In contrast, in depressed individuals with bipolar disorder, findings indicate bilateral reductions in both ventromedial prefrontal cortical-amygdala functional connectivity and fractional anisotropy in underlying white matter tracts.
These studies suggest that the depression of major depressive disorder, unlike bipolar depression, may be characterized more by left-sided than by bilateral abnormalities in implicit emotion regulation circuitry and underlying white matter tracts. This may be associated with reduced left prefrontal cortical activity during emotion processing in individuals with major depressive disorder (
151). Given the putative role of the left prefrontal cortex in processing approach-related emotions (
152), this bias away from left prefrontal cortical activity during emotion processing may result in the well-documented attentional bias away from positive and toward negative emotional stimuli (
153) and associated findings of abnormally increased amygdala (and anterior cingulate cortical) activity to negative emotional stimuli, described above. Links among these phenomena require further study, however. Bipolar disorder, by contrast, may be associated with bilateral dysregulation of the amygdala by different prefrontal cortical regions and may result in the emotional lability and abnormally elevated amygdala activity to both negative and positive emotional stimuli reported in individuals with bipolar disorder (
3). In support of this, one recent study showed a positive correlation between the magnitude of amygdala activity to positive emotional stimuli and levels of subthreshold manic symptoms in depressed individuals with major depressive disorder (
154).
Increasing evidence also suggests differential patterns of abnormalities in reward circuitry in individuals with major depressive disorder compared with those with bipolar spectrum disorders. For example, a recent review highlighted, in individuals with bipolar disorder across different mood states and different bipolar subtypes, abnormally elevated activity in the left ventrolateral prefrontal cortex, a region implicated in tracking reward value and arousal during anticipation of potentially rewarding stimuli (
155,
156), during anticipation of uncertain reward or uncertain losses (
3). This pattern of abnormal neural activity is not reported in individuals with current or remitted major depressive disorder (
9,
157). Given the role of the left prefrontal cortex in processing approach-related emotions (
152) and reports of heightened reward sensitivity in individuals with bipolar disorder (
158), elevated left ventrolateral prefrontal cortical activity may represent a neural marker of heightened reward sensitivity that distinguishes bipolar disorder from major depressive disorder.
Meta-analyses have also indicated reductions in hippocampal and striatal volumes in individuals with major depressive disorder relative to those with bipolar disorder (
113), which may be associated with greater amygdala activity to negative emotional stimuli, as described above, or may result from different patterns of psychotropic use in the two disorders (
3); further study is needed. Findings from resting-state studies directly comparing individuals with the different disorders are few and are difficult to interpret (
3). Overall, findings thus suggest that bipolar disorder may be distinguished from major depressive disorder by patterns of function and white matter structure in the two neural circuits of interest in this review.
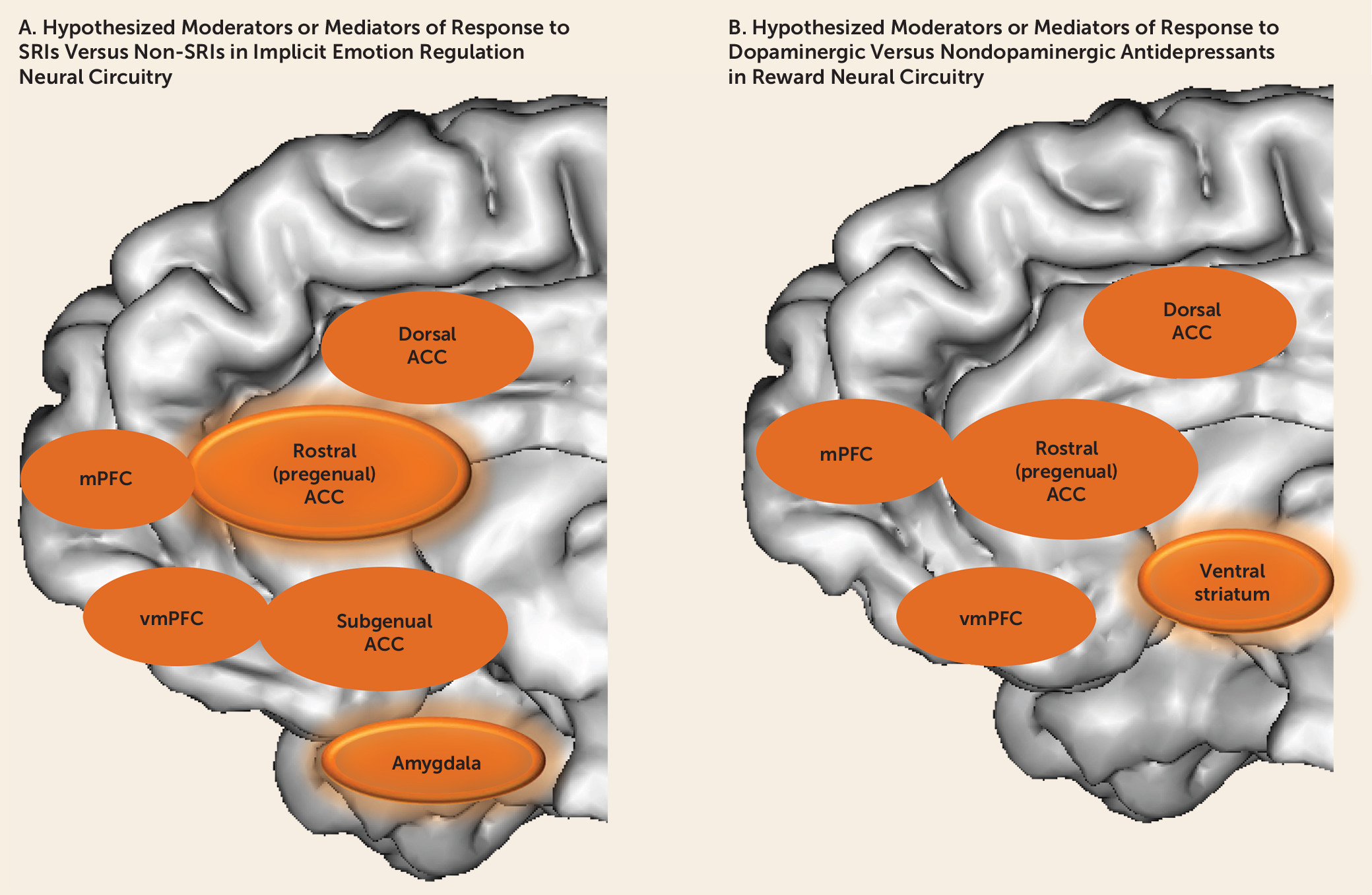
Despite the advances that neuroimaging techniques have provided in increasing our understanding of pathophysiologic processes in depression—specifically in implicit emotion regulation and reward circuits—the extent to which neuroimaging measures reflecting these processes moderate (and mediate) differential treatment response in individuals with depression remains understudied. An increasing number of small studies, however, have sought to identify neuroimaging predictors of treatment response in depression, as described in the following sections.
Future Directions for Neuroimaging Studies of Treatment Response Prediction in Depression
Clinical studies have traditionally made a choice between using large samples to test well-defined hypotheses and using smaller samples to allow in-depth assessment for hypothesis generating. The majority of neuroimaging studies, however, have focused on small samples with few assessments. Neuroimaging studies with large sample sizes are thus required for sufficient power to test key hypotheses and to subdivide data into training and testing data sets for first identifying and subsequently establishing moderators and mediators of treatment response. Furthermore, identifying, as early as possible after commencing treatment, measures that moderate and mediate treatment response remains a crucially important, but as yet unmet, need in clinical practice. Few studies have included neuroimaging assessments in early phases of treatment, and, of those that have (
6,
176), none have examined how such early changes in neuroimaging measures moderated or mediated subsequent treatment response. The inclusion of baseline and early-stage (e.g., 1 week after treatment onset) neuroimaging assessments and of more than one treatment in clinical trial platforms will help identify moderators and early mediators of differential treatment response, as opposed to focusing on predictors of successful response to a single treatment or to treatment in general. Additionally, while previous neuroimaging findings suggested a neural signature of placebo response (
177), no studies have examined the extent to which neuroimaging measures act as moderators or mediators of differential response to placebo compared with drug. Future studies should do so.
Studies would also benefit from examining more than one neural circuit, using multiple neuroimaging modalities, to examine the extent to which relationships among measures of the functional and structural integrity of parallel yet distributed neural circuits may moderate and mediate differential treatment response in depressed individuals. Here, the choice of medications in treatment platforms could include antidepressants, at various dosages, that would be expected to differentially affect function in serotonergically modulated implicit emotion regulation and dopaminergically modulated reward processing neural circuits (
Figure 2). These measures could be integrated with electrophysiological, neurocognitive, and clinical measures, using, for example, factor analysis, to identify key brain-behavior relationships that may moderate and mediate differential treatment response in depression (
178,
179). Finally, as in studies of cardiovascular disease, asthma, breast cancer, lung cancer, multiple sclerosis, macular degeneration, and other medical illnesses (
180–
184), future studies should identify personalized biosignatures developed from several clinical and biological markers reflecting underlying pathophysiologic processes. The combination of these approaches is more likely to be successful and to result in significant improvements in shorter- and longer-term clinical and functional outcome for the large number of individuals who suffer from depressive illnesses.