One of the most consistent and replicated postmortem findings in schizophrenia is the reduced expression of mRNA encoding the 67-kD isoform of glutamic acid decarboxylase (GAD67), the enzyme principally responsible for the synthesis of gamma-aminobutyric acid (GABA) (
1). The deficit in GAD67 mRNA appears to be prominent in the parvalbumin-containing subset of GABA neurons and to be conserved with similar magnitude across multiple cortical regions (
2). These findings may be interpreted as a reduced capacity for cortical GABA production. GABA neurotransmission plays a key role in sustaining synchronous oscillations in cortical networks (
3), which in turn is thought to be a critical neural mechanism for supporting a number of cognitive and perceptual processes (
4–
7). Thus, lower GABA synthesis in schizophrenia has been hypothesized to contribute to the altered prefrontal cortical oscillations and functional activation associated with impaired performance on working memory or cognitive control tasks (
8–
15), as well as to contribute to abnormalities in a range of other cognitive, affective, sensory, and motor functions that depend on GABA neurotransmission in a number of other cortical regions (
16–
19). However, to date, there is no direct, in vivo evidence that cortical GABA transmission is altered in schizophrenia or linked to cognitive and neurophysiological disturbances in this illness.
Results
The schizophrenia group included 10 African Americans and seven Caucasians; 11 of the group were male, and the mean age was 27.5 years (SD=6.8). The healthy comparison group included seven African Americans, 14 Caucasians, and one Asian; 13 of the group were male, and the mean age was 28.4 years (SD=8.7). There were no significant differences between groups on any demographic measure. The antipsychotic-naive group included five African Americans and three Caucasians; six of the group were male, and the mean age was 26.8 (SD=7.7). The antipsychotic-exposed group included five African Americans and four Caucasians; five of the group were male, and the mean age was 28.2 (SD=6.2). There were no significant differences between these subgroups on any demographic measure, nor between either of these groups and the healthy comparison group.
Participants’ scores on the PANSS and the MCCB are summarized in
Table 1.
PET Scan Parameters
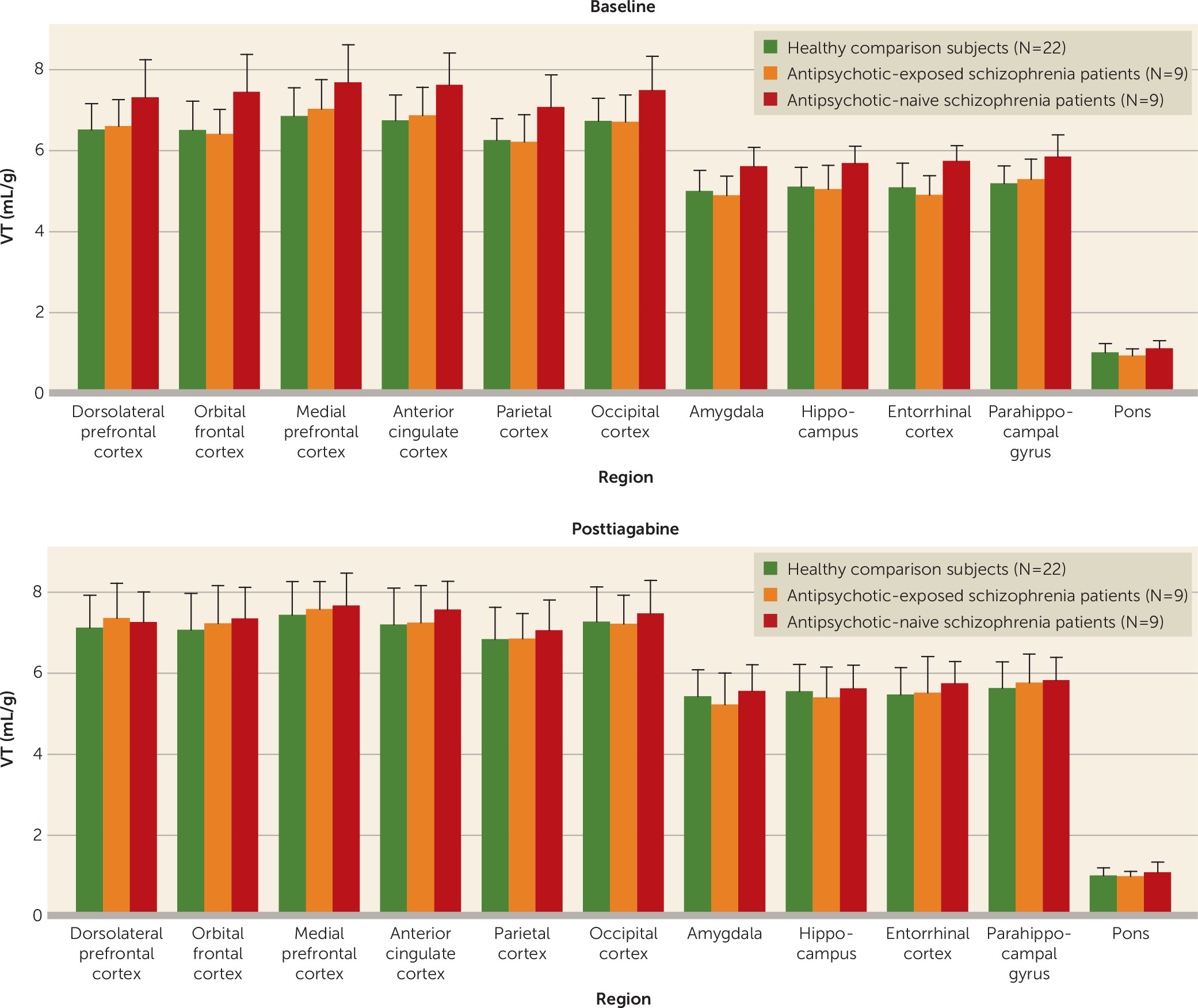
Neither the injected dose, specific activity, injected mass, free plasma fraction, nor V
ND differed between the baseline and posttiagabine scan for any of the groups (
Tables 2 and
3). Tiagabine administration resulted in a slight increase in the plasma clearance of [
11C]flumazenil in the healthy comparison group (
Table 2). No significant differences were detected between the schizophrenia and healthy comparison groups for either the baseline or the posttiagabine scan, with the exception of slightly higher injected mass in both conditions for the schizophrenia group (
Table 2). Comparison of the healthy comparison group with the antipsychotic-exposed schizophrenia group and with the antipsychotic-naive schizophrenia group revealed that the antipsychotic-exposed group had a higher injected mass than the healthy comparison group in both conditions (
Table 3). However, although numerically higher, the injected mass for all scans for all subjects remained within tracer dose range of <10 μg (
38) and would not be expected to affect the measurement of V
T. The free fraction was lower in the antipsychotic-naive schizophrenia group than in the antipsychotic-exposed group in the baseline condition, and lower than the healthy comparison group in both conditions (
Table 3).
Regional Distribution Volumes and Benzodiazepine Receptor Availability
Healthy comparison subjects.
Tiagabine administration significantly increased V
T in the large cortical regions, with a Bonferroni-corrected p value of 0.02 (
Table 4). Examination of V
T across the component regions of interest revealed a significant regional effect (F=151, df=12, 31, p<0.001), no region-by-condition interaction, and a significant difference across conditions (F=7.2, df=1, 42, p=0.01). On a region-by-region basis, significant increases in all regions were seen in the posttiagabine condition (
Table 4).
Schizophrenia patients.
No significant change in V
T was observed in any of the large cortical regions after administration of tiagabine (
Table 4). Examination of V
T across the component regions of interest revealed no significant difference across conditions with the omnibus test or with region-by-region contrasts (
Table 4).
Comparison of healthy comparison and schizophrenia groups.
No difference was observed when comparing [
11C]flumazenil ΔV
T between the healthy comparison and schizophrenia groups. Performing the repeated-measures ANOVA with the three groups (healthy, antipsychotic-exposed schizophrenia, and antipsychotic-naive schizophrenia) revealed a significant difference between groups (F=3.6, df=2, 33, p=0.04). Further analysis revealed that the antipsychotic-naive group had significantly lower ΔV
T compared with the healthy comparison group (F=5.93, df=1, 26, p=0.02). This difference reached significance in the dorsolateral prefrontal, orbital frontal, medial prefrontal, and parietal cortices (
Table 5). No significant differences were observed in [
11C]flumazenil ΔV
T between the healthy comparison and antipsychotic-exposed schizophrenia groups or between the antipsychotic-naive and antipsychotic-exposed schizophrenia groups.
To examine the proximal source of the low ΔV
T in the antipsychotic-naive schizophrenia group, the baseline and posttiagabine V
T values were compared with those of the healthy comparison group and the antipsychotic-exposed schizophrenia group. The repeated-measures ANOVA across the three groups on baseline [
11C]flumazenil V
T revealed a significant difference between groups (F=5.81, df=2, 36, p=0.007), with the antipsychotic-naive schizophrenia group demonstrating elevated baseline [
11C]flumazenil V
T compared with the healthy comparison group (F=10.39, df=1, 28, p=0.003) and the antipsychotic-exposed group (F=6.68, df=1, 15, p=0.02). No differences in posttiagabine [
11C]flumazenil V
T were observed in the initial analysis or when the groups were compared separately (
Table 6 and
Figure 1). No differences were observed when comparing [
11C]flumazenil binding in the healthy comparison group and the overall schizophrenia group at baseline or after tiagabine administration, or between the healthy comparison group and the antipsychotic-exposed group in baseline or posttiagabine [
11C]flumazenil V
T.
Given the finding of elevated [11C]flumazenil VT at baseline in the antipsychotic-naive schizophrenia group, we examined the relationship between time off medications in the antipsychotic-exposed schizophrenia subjects and [11C]flumazenil VT at baseline to see whether treatment had an effect on this measurement. A correlation was seen between weeks off of medications and baseline [11C]flumazenil VT in the medial temporal lobe, although it did not meet the Bonferroni-corrected significance threshold of p<0.02 (r=0.73, p=0.026).
Clinical and Cognitive Measures
PANSS positive score was correlated with [11C]flumazenil ΔVT in the medial temporal lobe (r=0.76, p=0.02) and the medial temporal lobe subregion of the amygdala (r=0.88, p=0.002) in the antipsychotic-naive schizophrenia group, using a Bonferroni-corrected p<0.02 for the medial temporal lobe functional region and p<0.005 for the amygdala (see Figure S2 in the online data supplement). No correlations were noted for [11C]flumazenil ΔVT with PANSS score or subscores in the antipsychotic-exposed schizophrenia group or the schizophrenia group as a whole. Baseline [11C]flumazenil binding in the antipsychotic-exposed group was negatively correlated with PANSS positive score in the orbital frontal cortex (r=−0.80, p=0.009), but the correlation did not survive Bonferroni correction (p<0.005). No other correlations were observed between [11C]flumazenil binding (baseline, posttiagabine, or ΔVT) and PANSS measures.
Baseline [11C]flumazenil VT in the antipsychotic-naive schizophrenia group, but not in the antipsychotic-exposed group or the schizophrenia group as a whole, was negatively correlated with the visual learning cognitive domain of the MCCB (using a Bonferroni-corrected threshold of p<0.02 for the functional regions and p<0.005 for the subregions). This negative correlation was seen in the medial temporal lobe (r=−0.74, p=0.02) as well as in one of its subregions, the entorhinal cortex (r=−0.87, p=0.002). In the antipsychotic-naive schizophrenia group, other regions had similar negative correlations, albeit falling short of significance, between baseline [11C]flumazenil VT and the MCCB visual learning domain. In addition, negative correlations with baseline [11C]flumazenil VT, also falling short of significance, were observed across several of the other MCCB domains in various regions in the antipsychotic-naive schizophrenia group but not in the antipsychotic-exposed group or the schizophrenia group as a whole.
Cortical Oscillations
For the healthy comparison group, the association between gamma-band power and the ability to increase extracellular GABA levels was significant in the large cortical area of the association cortex (r=0.69, p=0.04; see Figure S3 in the online data supplement) and the dorsolateral prefrontal cortex (r=0.69, p=0.04), although these relationships did not survive Bonferroni correction. No association between [11C]flumazenil ΔVT and gamma-band power was noted in the schizophrenia group as a whole or in the antipsychotic-exposed and antipsychotic-naive groups.
In the antipsychotic-naive group, but not in the antipsychotic-exposed group, the schizophrenia group as a whole, or the healthy comparison group, gamma-band power was strongly correlated with baseline [11C]flumazenil VT in the medial temporal lobe (r=0.94, p<0.001), the association cortex (r=0.78, p=0.01), and the sensory cortex (r=0.79 p=0.01) (see Figure S4 in the data supplement).
Discussion
After acute GAT1 blockade in healthy subjects and in antipsychotic-naive and antipsychotic-exposed schizophrenia patients, we detected the predicted elevated extracellular GABA levels as increased binding of the benzodiazepine receptor ligand, measured as [11C]flumazenil ΔVT, in the healthy comparison group. However, schizophrenia patients did not exhibit this same increase in [11C]flumazenil VT after GAT1 blockade, indicating impaired GABA transmission in this population. Antipsychotic-naive schizophrenia patients showed an absence of change in [11C]flumazenil binding after acute increase in GABA and increased baseline [11C]flumazenil binding, indicating that this subgroup contributed disproportionally to the group effect. In contrast, schizophrenia patients who had past treatment with antipsychotics were indistinguishable from healthy comparison subjects on the PET scan measurements.
There are two potential interpretations of the finding of increased [11C]flumazenil binding at baseline with no change in binding after tiagabine administration in antipsychotic-naive schizophrenia patients. The first possibility is that elevated [11C]flumazenil binding at baseline reflects greater affinity due to higher extracellular GABA levels prior to tiagabine exposure. Consequently, tiagabine administration does not result in significant additional elevations of synaptic GABA in this population. Alternatively, elevated baseline [11C]flumazenil binding may indicate a compensatory increase in GABAA receptors in response to a deficit in GABA transmission early in the illness, with lower presynaptic GABA synthesis limiting the effect of tiagabine on extracellular GABA levels.
The postmortem findings of lower levels of GAD67 mRNA and protein, which are most prominent in parvalbumin-containing interneurons, as well as alterations in other markers of GABA function, suggest that impairment in inhibitory neurotransmission could contribute to the symptoms of schizophrenia (see reference
1 for a review). However, reductions in GAD67 mRNA and protein do not, in and of themselves, support lower GABA levels, as these findings could result from a down-regulation of GAD67 transcription in parvalbumin neurons in response to elevated GABA levels. Furthermore, lower GABA synthesis in parvalbumin neurons does not exclude the possibility of other interneurons releasing greater-than-normal levels of GABA (
39) in response to elevated activity of excitatory pyramidal neurons, as proposed in the
N-methyl-
d-aspartate receptor hypofunction hypothesis of schizophrenia (
40). Thus GABA may be elevated globally to maintain homeostasis in the face of perturbations in circuit activity. Since GAT1 is widespread across the neocortex and present in both GABA neurons and glial cells (
41), the technique employed in our study is well suited to detect alterations in global extracellular GABA levels, but it does not have the resolution to detect localized, circuit-specific perturbations in GABA levels.
Similarly, the other brain imaging technique used to measure GABA levels in vivo, magnetic resonance spectroscopy (MRS), provides a measurement of global GABA levels generated from the average tissue concentration of GABA in all pools (intra- and extracellular), as opposed to PET, which allows the detection of alterations in GABA levels in the extracellular space. Of the published MRS studies exploring GABA in schizophrenia, three demonstrated increased GABA measures in schizophrenia (
42–
44), three demonstrated decreased GABA measures (
45–
47), and three showed no difference (
42,
48,
49). Two of the MRS studies examined the effect of treatment with antipsychotic medications; Kegeles et al. (
42) found increased GABA measures in the medial prefrontal cortex and normal GABA levels in the dorsolateral prefrontal cortex in unmedicated patients, with no effect of previous treatment. Tayoshi et al. (
49) found that the lower the dosage of antipsychotic medication, the higher the GABA levels in the anterior cingulate cortex. Both of these findings are consistent with one interpretation of our findings, that basal GABA levels are elevated in antipsychotic-naive schizophrenia patients. They are also consistent with the observed association (falling short of significance) between time off antipsychotic medications and baseline [
11C]flumazenil binding in antipsychotic-exposed schizophrenia patients.
Alternatively, our findings could be viewed as reflecting a compensatory increase in GABA
A receptors in response to a deficit in GABA transmission early in the illness, such that a lower pool of presynaptic GABA limits the effect of tiagabine on extracellular GABA levels. We believe this second interpretation is more likely to be the case, for the following reasons. First, this interpretation is consistent with postmortem studies showing increases in GABA
A receptor binding (
50) in schizophrenia. Although findings from postmortem studies of benzodiazepine receptors have been mixed—reporting no change (
51), decreases (
52), or increases (
53)—these findings of variable differences in benzodiazepine binding could be due to differences in previous treatment, and if so, they are in line with the differences we observed in relation to the effects of the presence or absence of previous treatment on [
11C]flumazenil binding at baseline, as the postmortem studies do not report on antipsychotic-naive individuals. Previous imaging studies of benzodiazepine receptor densities in schizophrenia have found no differences (
26,
27,
54–
56). Only the study by Asai et al. (
56) reported on medication-naive individuals separately and found no difference; however, the study used the pons as a reference region and used [
11C]Ro15-4513, a ligand that measures separate, albeit overlapping, populations of benzodiazepine receptors (
24), making direct comparison with our results difficult. Second, in our study, schizophrenia patients who had never received antipsychotic treatment had elevated benzodiazepine binding, whereas in those who had received treatment, benzodiazepine binding was no different from that in healthy subjects, with a notable association, approaching significance, between time off medication and benzodiazepine binding. These findings may be explained by low GABA transmission in the illness initially, with a compensatory increase in GABA
A receptors. Subsequent treatment with antipsychotics may increase GABA transmission, resulting in normalization of GABA
A receptor levels, perhaps through a reduction of excess dopamine D
2 receptor stimulation at the convergence of cortical glutamatergic afferents and dopamine projections on GABA-ergic medium spiny neurons (
57), although some preclinical work suggests that antipsychotic medications reduce GABA transmission (
58). However, continued impairment in other, as yet unknown, processes prevents this normalization from being effective in overcoming the deficits in cognition.
Third, the lack of correlation between gamma power and [
11C]flumazenil ΔV
T in the schizophrenia group but not the healthy comparison group further supports impaired GABA neurotransmission in the illness. Consistent with the hypothesis that GABA transmission is critical for various types of perceptual and cognitive processes, in the antipsychotic-naive schizophrenia group, we observed negative correlations between the visual learning cognitive domain of the MCCB and baseline [
11C]flumazenil binding (again, interpreted as increased in response to reduced GABA transmission). This association between impaired GABA transmission and impaired cognition in schizophrenia is supported by experimental models (
59) that suggest that GABA
A receptor-mediated transmission is required for the induction of network oscillations. In turn, synchronous gamma activity has been proposed to be critical for perceptual feature binding and to be associated with higher cognitive processes (
4,
60). We measured oscillatory activity during a cognitive load in an attempt to directly link the measurement of GABA in vivo with this phenomenon in our subjects (see the
online data supplement). We found no association between GABA transmission and the ability to increase oscillatory activity in the gamma-band range in the schizophrenia group as a whole or when broken down to the antipsychotic-exposed and antipsychotic-naive groups. Interestingly, we noted a strong relationship between baseline [
11C]flumazenil binding in antipsychotic-naive schizophrenia patients and gamma-band power, perhaps indicating that a compensatory increase in GABA
A receptors is effective in increasing the ability to entrain cortical networks, albeit not to a sufficient degree to improve cognitive performance. Alternatively, baseline GABA increases could result from increased baseline activity of parvalbumin-positive interneurons, which in turn could give rise to network gamma oscillations (
61). In fact, increases in baseline gamma activity have been reported in schizophrenia and have been invoked to explain decreases in task-activated gamma activity, given the standard practice of subtracting prestimulus baseline activity (
62). However, on this account, the increases in baseline [
11C]flumazenil binding would negatively correlate with task-activated gamma. In other words, a higher baseline gamma activity, resulting from increased baseline GABA, would result in decreased task related-activated gamma when subtracting prestimulus activity from the task-activity measure, meaning that subjects with higher baseline GABA (i.e., higher prestimulus activity) would have the lower gamma, contrary to our findings of a positive relationship.
Our finding of a relationship between clinical symptoms and markers of GABA-ergic transmission is consistent with and extends previous findings. Although it did not survive correction for multiple comparisons, the negative correlation between PANSS positive symptom score and baseline [
11C]flumazenil V
T in the orbital frontal cortex is consistent with the findings reported by Busatto et al (
26); however, Schröder et al. (
27) found a
positive correlation with the total score on the Brief Psychiatric Rating Scale. The finding of a positive relationship between [
11C]flumazenil ΔV
T and PANSS positive symptom score in the antipsychotic-naive but not the antipsychotic-exposed schizophrenia group is difficult to interpret in the context of a minimal change in V
T in the antipsychotic-naive group and a lower degree of positive symptoms in the antipsychotic-exposed group; further studies are necessary to explore this finding. Interestingly, we did not observe any relationship between negative symptoms and [
11C]flumazenil binding parameters, in contrast to the report of Asai et al. (
56), who reported a negative correlation between benzodiazepine binding and PANSS negative symptom score.
Taken together, the results of this study are consistent with postmortem studies suggesting lower cortical GABA neurotransmission in schizophrenia (see reference
1 for a review). Our data indicate that impairment in GABA transmission and reduced GABA signaling are most pronounced prior to treatment; with treatment, the abnormalities in the receptor parameters and GABA transmission measured by this paradigm appear to normalize.
The strengths of this study include measurement of the arterial input function, allowing for the assessment of the effects of tiagabine on V
ND and free plasma fraction. While the absence of change in these variables after tiagabine administration validates the use of either BP
P or BP
ND as an outcome measure, we chose to use V
T as our primary outcome measure. We were concerned that differential effects of increasing GABA levels on [
11C]flumazenil-specific binding in the pons would have an effect on the comparison across groups. Differences in specific binding within the pons would affect either BP
P or BP
ND to a greater degree in one group relative to the other, potentially obscuring group differences in tiagabine-induced change in [
11C]flumazenil binding, despite the fact that, on average, no changes were seen in the pons V
T after tiagabine administration in either group. Moreover, V
T has been shown to be a more reliable and robust outcome measure for [
11C]flumazenil than either BP
ND or BP
P (
37).
This study also has several limitations, among which is the fact that only minimal information on total exposure to antipsychotic medications was available for the previously treated group, thereby limiting our ability to explore the relationship between our outcome measures and time and type of medication. In addition, our previously published studies noted a high variability in the percent change in [
11C]flumazenil binding across subjects (
20,
21). The present study was consistent with this, as we again saw a high degree of variability in [
11C]flumazenil ΔV
T across all of the regions of interest. Detecting differences between individuals with a psychiatric disorder and healthy comparison subjects remains challenging with this level of variability; however, comparing the groups in this study, we noted the increase in [
11C]flumazenil V
T to be greater in the healthy comparison group than in the antipsychotic-naive schizophrenia group, in which there was a near absence of tiagabine-induced increase in [
11C]flumazenil V
T. In other words, this relatively large between-group difference could be detected with the present method (the average increase in V
T in the healthy comparison group was 8.3% [SD=2.2%], whereas it was −0.51% [SD=1.1%] for the antipsychotic-naive schizophrenia group), but more subtle differences in GABA availability between the antipsychotic-exposed group and the healthy comparison or antipsychotic-naive group may be difficult to detect without improvements in the methods to reduce the variability.
The results of this study suggest that GABA abnormalities in schizophrenia are widespread across cortical domains, consistent with recent postmortem studies (
2), and are linked to clinical symptoms and cognitive impairments of the illness. In addition, treatment with antipsychotic medications appears to normalize the measured abnormalities in GABA signaling; however, the clinical impact of this normalization appears minimal at best with regard to cognitive functioning.