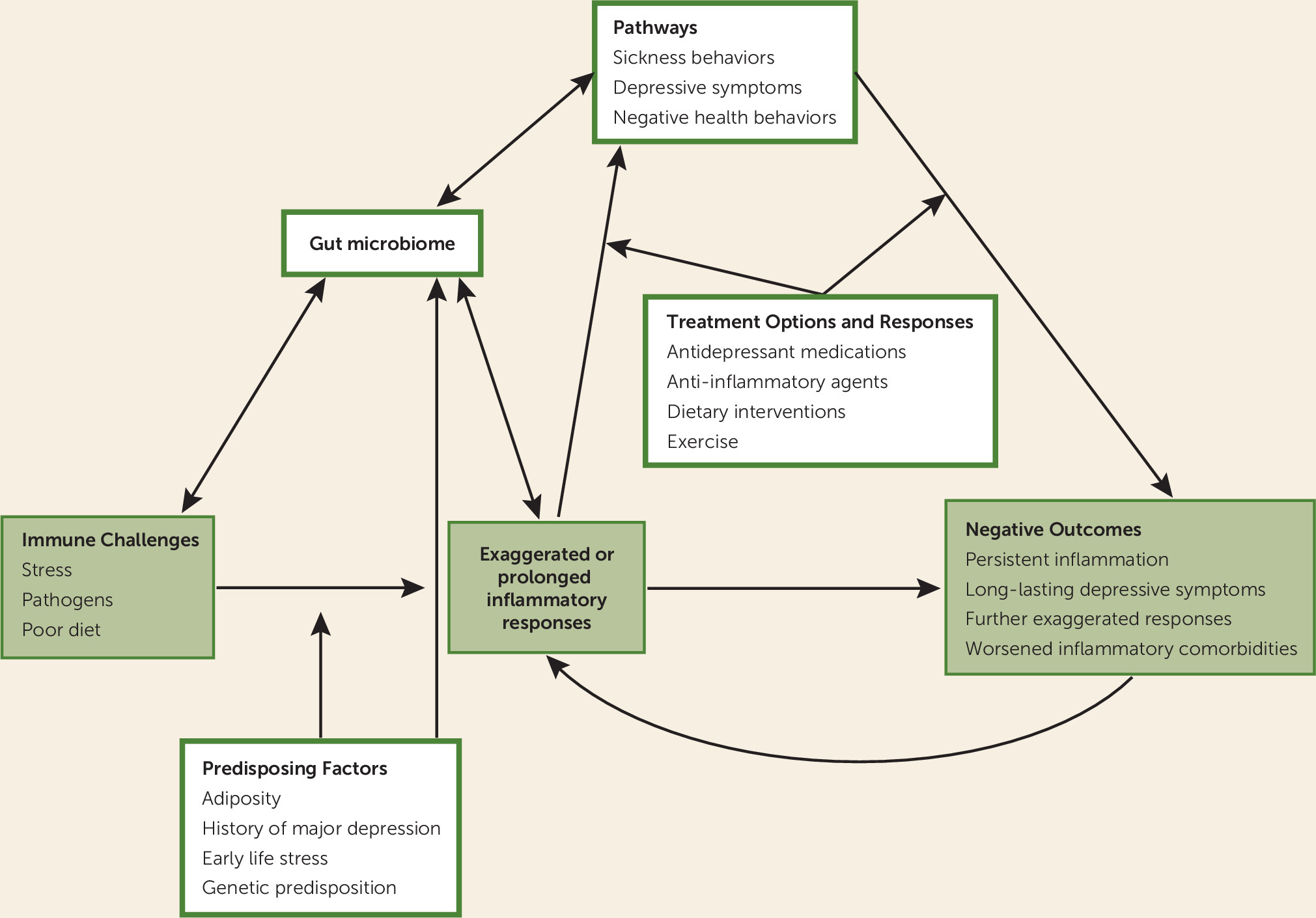
Depression and inflammation are intertwined, fueling and feeding off each other. This bidirectional loop, in which depression facilitates inflammatory responses and inflammation promotes depression, has clear health consequences. Heightened inflammation characterizes a number of disorders and systemic diseases, including cardiovascular disease, diabetes, metabolic syndrome, rheumatoid arthritis, asthma, multiple sclerosis, chronic pain, and psoriasis; each of these also features an elevated risk for depression (
1,
2).
Three meta-analyses have highlighted proinflammatory cytokine differences between patients with major depressive disorder and controls, including interleukin-6 (IL-6), tumor necrosis factor-alpha (TNF-α), IL-1β, the soluble IL-2 receptor, the IL-1 receptor antagonist (IL-1ra), and C-reactive protein (CRP) (
3–
5). The stronger associations in clinical samples compared with community samples provide evidence of dose-response relationships (
3). Supporting a causal pathway, higher IL-6 and CRP predicted the subsequent development of depressive symptoms (
6). Relatedly, prospective studies also showed that depression predicted later IL-6 and CRP (
7–
10).
The pediatric literature also demonstrates bidirectional pathways between inflammation and depression (
11). Data from two population-based prospective studies provided evidence for depression-inflammation relationships early in life. Children with higher IL-6 levels at age 9 were more likely to be depressed at age 18 compared with those with low IL-6 levels; importantly, IL-6 was measured prior to depression onset, thus suggesting that high IL-6 is indeed a risk factor (
12). In another study with children who were 9, 11, or 13 years old at intake, depression predicted subsequent CRP level, with higher CRP levels following multiple depressive episodes (
7).
However, depression is complex, and inflammation may contribute only in a subpopulation. Data from the National Health and Nutrition Examination Survey provide a rough estimate of the prevalence of heightened inflammation in depressed people; 47% of those whose depression inventory scores were above the clinical threshold had a CRP level ≥3.0 mg/L, and 29% had a CRP level ≥5.0 mg/L (
13). Raison and Miller (
14) suggest that inflammatory markers are noticeably higher in about a third of depressed patients compared to the majority of nondepressed comparison subjects. Thus, inflammation is neither necessary nor sufficient to induce or sustain depression (
14,
15), but it clearly plays an important role in a substantial subpopulation (
16). It follows that positive clinical responses to anti-inflammatory interventions may only occur in the subset with heightened inflammation (
17,
18). Accordingly, we address mechanistic pathways between depression and inflammation, and then turn to questions of how (pathways) and for whom (predispositions) these links exist, with a focus on integrating newer research relevant to depression initiation, treatment response, and risk for recurrence (
Figure 1).
Mechanistic Pathways
Cytokines induce depressive symptoms by influencing diverse mood-related processes. Elevated inflammatory signaling dysregulates neurotransmitter metabolism, impairs neuronal health, and alters neural activity in mood-relevant brain regions (
2,
19).
Peripherally released cytokines send signals via molecular, cellular, and neural routes, which ultimately reach the brain and enhance CNS inflammation (
2,
19,
20). Cytokines alter production, metabolism, and transport of neurotransmitters that synergistically affect mood, including dopamine, glutamate, and serotonin (
21). For example, cytokines stimulate indoleamine 2,3-dioxygenase, an enzyme that affects tryptophan metabolism. This well-established pathway promotes depression by simultaneously slowing serotonin production and enhancing levels of kynurenine, a tryptophan metabolite (
22).
Inflammation also affects neuronal growth and survival. Cytokines contribute to oxidative stress, which damages glial cells in mood-relevant brain regions, such as the prefrontal cortex and the amygdala (
23). Cytokine-induced glutamate dysregulation can lead to excitotoxicity, thereby decreasing production of neurotrophic factors (e.g., brain-derived neurotrophic factor, BDNF) that typically support neuronal health, neuroplasticity, and neurogenesis (
24). Notably, these neurotransmitter and cellular changes alter brain activity and neurocircuits underlying distress, motivation, and motor function (
16,
19).
In addition to their effects on neural processes, cytokines promote dysregulated hypothalamic-pituitary-adrenal (HPA) axis functioning, a key characteristic of depression (
25,
26). Abnormal glucocorticoid signaling can influence the maintenance and progression of depression (
27). Briefly, glucocorticoids typically dampen inflammation via a negative feedback loop. However, inflammation can cause glucocorticoid resistance in immunocytes and their cellular targets by inducing MAP kinases c-jun
N-terminal kinase (JNK) and p38 (
1). In this way, cytokine signal transduction pathways (e.g., nuclear factor-kB, NF-kB) disrupt glucocorticoid receptor function and expression, leading to unrestrained inflammatory responses that could further fuel depressive symptoms (
2,
26,
28). Cytokine-dependent glucocorticoid receptor resistance decreases inhibitory feedback on production of corticotropin-releasing hormone and cytokines, intensifying the stress-response system (
29). The glucocorticoid receptor protein is abundantly expressed throughout the main neuronal subregions of the hippocampus. BDNF functions as a powerful modulator of structural plasticity in the hippocampus and mediates protective influences by enhancing neuronal survival (
30). Sustained glucocorticoid exposure leads to dendritic atrophy in hippocampal subfields and decreases neuronal cell survival by evoking a decline in BDNF expression in hippocampal and cortical regions (
31,
32).
Inflammation may affect people differently, depending on their individual physiology; some people have bodily systems that protect them from developing inflammation-based depression, while others do not. Mechanistically, even lower levels of inflammation could be depressogenic in vulnerable individuals; Raison and Miller (
14) call this phenomenon “immune response element amplification.” These may include lower parasympathetic activity, poorer sensitivity to glucocorticoid inhibitory feedback, lower BDNF production, larger responses to social threat in the anterior cingulate cortex or amygdala, and smaller hippocampal volume. Indeed, these are all correlates of major depression that would influence sensitivity to the depressogenic consequences of inflammatory stimuli.
Traveling Companions: Inflammation and Depression
The bidirectional links between inflammation and depression have received considerable attention (
1,
2,
16,
33–
36). Heightened inflammation alerts the CNS to induce or intensify “sickness behaviors,” including negative mood, fatigue, anhedonia, increased pain sensitivity, loss of appetite, and cognitive deficits, a cluster of symptoms resembling human depression (
29,
34,
37). For example, administration of cytokines, endotoxin, or vaccines has been found to worsen mood, fatigue, and pain sensitivity and to boost proinflammatory cytokine production in healthy volunteers (
34).
Inflammatory mediators can also induce clinical depression, bolstering support for inflammation’s role in depression’s pathophysiology. These effects can be substantial; for example, cytokine therapies, used for treating some cancers and chronic viral infections, provoke the onset of major depression in up to 45% of patients (
21,
38–
40). Most people who receive interferon-alpha (IFN-α) treatment develop neurovegetative symptoms, including fatigue, sleep problems, anorexia, and psychomotor retardation; these symptoms persist throughout treatment (
21). However, mood and cognitive symptoms develop primarily in vulnerable patients, including those with a mood disorder history or higher initial levels of depressive symptoms, chronic inflammatory exposure, higher baseline levels of inflammation, or genetic polymorphisms associated with risk for depression or inflammation (
21,
38,
39).
Antidepressant medication responsiveness may be poorer in patients with major depression who have heightened plasma inflammatory markers, as well as those with polymorphisms in inflammation-related genes and proinflammatory gene expression profiles (
17,
41–
51). Recently, a provocative trial (
17) assessed the efficacy of the monoclonal antibody infliximab, a TNF-α antagonist, in 60 patients with major depression that was at least moderately medication resistant. Despite the absence of any overall benefit for infliximab versus placebo, patients with high baseline CRP levels had substantially greater reductions in depressive symptoms than those with low CRP levels.
The Gut Microbiota, Inflammation, and Depression
The gut-brain axis involves bidirectional communication between the CNS and the gastrointestinal tract via neurocrine and endocrine signaling pathways (
52). Physical and psychological stressors can alter the gut microbiota’s composition and metabolic activities, and signals produced by the gut microbiota can in turn affect the brain and emotional responses (
52). Alterations in the gut microbiota shape physiology through contributions to inflammation, obesity, and mood, among other things (
53). For example, both rodent and human studies provide causal evidence linking obesity and the gut microbiome (
53).
Depression can promote intestinal permeability, that is, greater inflammation-inducing endotoxin translocation, described as a “leaky gut.” Indeed, depressed patients have been found to have higher antibody against gut bacteria than comparison subjects (
54). In another study, patients with major depression had elevated expression of 16S rDNA, a marker of bacterial translocation, compared with nondepressed comparison subjects, and the magnitude was correlated with depressive symptom severity (
55). Among alcohol-dependent patients, those with higher depression, anxiety, and craving symptom ratings also had greater gut permeability and gut-bacterial dysbiosis than those with normal gut permeability (
56).
Targeting the gut-brain axis may offer novel treatment options with benefits mediated through the vagus nerve, spinal cord, or neuroendocrine system (
57). Diet plays a key role in the gut’s microbiota composition and thus represents one potential therapeutic avenue, as do supplements (particularly probiotics and prebiotics) and medications, including antibiotics (
52). In rats, probiotic pretreatment attenuated gut leakiness after a restraint stressor (
58). Limited data from human subjects suggest that selected probiotics may reduce depressive symptoms as a result of their anti-inflammatory properties as well as their ability to reduce HPA axis activity (
57).
Rodent studies show how the microbiota’s composition has potent effects on brain biochemistry and behavior early in development (
53). Early-life maternal separation in mice can produce both long-lasting changes in HPA stress responses as well as persistent microbiome alterations (
57), evidence for one pathway through which early adversity induces depression and inflammatory responses in adults.
Early Adversity
Adults who experienced abuse or neglect as children are more likely to develop psychiatric disorders (
59). Indeed, childhood maltreatment is a particularly potent risk factor for depression in adults, especially when individuals encounter stressful life events (
2,
59). Early adversity also predicts a greater risk for recurrent, treatment-resistant depressive episodes (
59).
Convergent evidence shows that childhood adversity can have longer-term inflammatory consequences (
60–
64). Among adults with major depression, those with a history of early maltreatment had higher CRP levels than those without such a history (
60). Additionally, early-life adversity was still associated with heightened IL-6 and TNF-α in an older adult sample with a mean age of 70 (
61). Adult survivors of childhood abuse also have maladaptive alterations in the HPA axis and autonomic stress responses compared with similar individuals without an abuse history (
65). For example, those with a history of early-life stress have lower heart rate variability, reflecting lower parasympathetic activity (
62), which is linked to inflammation. Trauma survivors have enhanced glucocorticoid resistance and increased central corticotropin-releasing factor activity, further supporting neuroendocrine stress response sensitization in those with early adversity (
65). Furthermore, inflammation-relevant epigenetic alterations associated with early adversity include alterations in glucocorticoid receptor expression (
62).
Early adversity can enhance inflammatory responsiveness to stressors. IL-6 levels rose higher after a laboratory stressor in individuals who reported childhood trauma compared with those without a trauma history (
66). These laboratory stress data parallel differences observed in response to daily stressors: IL-6 levels were 2.35 times greater in individuals with a childhood abuse history who experienced multiple stressors in the past 24 hours compared with participants with multiple daily stressors but no abuse history (
67).
Sickness Behaviors: Paving the Way to Inflammation and Depression
Sickness behaviors serve an adaptive function by conserving energy during an acute illness (
1). However, these symptoms can in turn fuel inflammation and depression, and thus it is not surprising that they can also predict treatment resistance and poorer treatment outcomes (
68).
Pain generates an inflammatory response (
69,
70), and amplified pain sensitivity serves as an additional inflammatory source that in turn provokes depressive symptoms (
71,
72). The association appears to be reciprocal: greater pain is associated with a higher prevalence of depression, and improvements in depression are correlated with declines in pain (
73). Pain increases the risk for recurrence of depression by worsening subthreshold depressive symptoms (
74). Greater pain severity is associated with poorer treatment outcomes in depression, including poorer responses to antidepressant medications (
73,
74).
Disturbed sleep, a cardinal symptom of depression, also has a contributory role, producing a twofold higher risk for depression (
75). Sleep loss stimulates production of proinflammatory cytokines and cellular inflammatory signaling, thus facilitating depression (
75). In turn, heightened inflammation disrupts sleep regulation (
76,
77); pharmacological cytokine blockers can normalize sleep (
75). In a longitudinal study, sleep disturbances increased the risk for systemic inflammation at the 5-year follow-up (
78).
Thus, sleep and pain are additional, independent accelerators for depression and inflammation that also act in tandem, building on each other. Disturbed sleep exacerbates pain and fatigue (
79). Conversely, pain clearly impairs sleep (
79). Changes in appetite, another key symptom of both inflammation and depression, can be triggered by sleep loss and fatigue (
76,
79). The poorer mood-related dietary choices that typically follow serve to promote inflammation and depression.
Diet as a Road to Depression and Inflammation
Observational studies have linked healthier diets with a lower risk for depression (
80,
81). Prospective studies suggest that healthier diets offer some protection against the development of both depressive symptoms and depressive disorders (
82,
83).
In addition to altering the risk for depression, diet quality also influences inflammation. Patients with metabolic syndrome (
84) who were randomly assigned to a Mediterranean-style diet for 2 years had significant reductions in CRP and IL-6 levels. In a twin study (
85), adherence to a Mediterranean diet was associated with lower IL-6 levels, and the results were not a function of shared environmental variance or genetic factors.
An innovative prospective study (
86) addressed the question of whether a Mediterranean-style diet lowered the risk of increased inflammation over time in older adults with depressive symptoms at study entry. At the 6-year follow-up, the average increase in IL-6 levels was larger in depressed participants who had not followed a Mediterranean-style diet than in all other groups; in contrast, IL-6 levels did not change in those who were depressed but followed a Mediterranean-style diet, suggesting that the healthier diet buffered the impact of depression on inflammation (
86).
To assess the question of whether inflammation serves as a mediator between diet and depression, researchers employed an empirically derived inflammatory dietary pattern score related to CRP, IL-6, and TNF-RII (
87). Using food frequency questionnaire data collected six times over 18 years in the large Nurses’ Health Study, they found that the risk for depression increased with higher inflammatory scores in women who were not depressed at baseline (
87).
Along with diet quality, quantity and timing matter. Caloric restriction produces powerful anti-inflammatory effects over periods of months to years (
88). Intriguingly, caloric restriction is also strongly antidepressant in rodent depression models (
89).
Even intermittent fasting or time-restricted feeding can reduce inflammation. Comparisons of IL-6 and CRP in observant Muslims 1 week before the month of Ramadan (no eating or drinking during daylight), during the final week, and 20 days after Ramadan showed that daytime fasting decreased IL-6 and CRP levels by about 50% compared with pre-Ramadan values, a dramatic reduction in the absence of weight change
; a nonfasting group assessed at the same times showed no IL-6 or CRP changes (
90). Time-restricted feeding also reduced inflammation in mice (
91). Additionally, TNF-α and IL-1ra responses to endotoxin were attenuated in rats that fasted for 48 hours compared with nonfasted rats (
92).
Short-term fasting can also benefit mood. Clinical observational studies have reported reductions in depressive symptoms that appear between days 2–7 of fasting (
93). Accordingly, anorexia may serve an adaptive function in both clinical depression and inflammation-induced sickness behavior by reducing inflammation (
94).
Thus, cross-sectional, prospective, and randomized-controlled-trial research demonstrates how diet quality, quantity, and timing influence both depression and inflammation. Diet-related inflammation can promote depression, and diet-linked depression in turn heightens inflammation. One dietary component, fish oil, has generated considerable interest.
Omega-3 Fatty Acids
Fish oil is the prime source for two key omega-3 fatty acids, eicosapentaenoic acid (EPA) and docosahexanoic acid (DHA). Patients with depression have, on average, lower plasma levels of omega-3 fatty acids than nondepressed comparison subjects. Furthermore, there are relationships within these populations between depressive symptom severity and omega-3 fatty acid plasma levels (
95,
96).
Five meta-analyses of randomized controlled trials have reached different conclusions about the efficacy of omega-3 fatty acids for treatment of depression. The first concluded that omega-3 fatty acid supplementation benefited clinically depressed individuals, but not those with less severe depressed mood (
97). In contrast, a second that focused only on major depression found that omega-3 fatty acids had a small, nonsignificant effect (
98). A third determined that omega-3 fatty acid supplementation was effective in both patients with major depression and those with subclinical depressive symptoms (
99). Two further meta-analyses suggested that EPA, not DHA, was the key omega-3 fatty acid related to efficacy in treating depression (
100,
101), consistent with the evidence for EPA’s stronger anti-inflammatory properties compared with DHA (
102,
103).
Epidemiological and observational studies have demonstrated that lower omega-3 fatty acid levels are associated with higher serum IL-6, TNF-α, and CRP (
104–
106). In contrast, most randomized controlled trials have not produced reliable serum cytokine changes (
107); the strongest support for the anti-inflammatory properties of omega-3 fatty acids in vivo has come from studies with older, hypertriglyceridemic or diabetic individuals with elevated inflammatory markers (
102), as well as a randomized controlled trial with sedentary, overweight middle-aged and older adults (
108).
However, inflammatory challenge studies provide compelling evidence of protective effects. Omega-3 fatty acids have been shown to attenuate both endotoxin and IFN-α-induced inflammation and sickness behavior in rodents and humans (
109–
113). In a randomized controlled trial in which patients received EPA, DHA, or placebo for only 2 weeks before initiation of IFN-α treatment, EPA significantly reduced the incidence of IFN-α-induced depression, and both EPA and DHA substantially delayed onset of major depression (
42). EPA was more effective than DHA, consistent with two meta-analyses (
100,
101). Importantly, the study population was subjected to an inflammatory insult that carried a high risk for depression, providing a backdrop that highlighted the reduction in risk.
Both the results of inflammatory challenge studies and meta-analyses suggest that heightened pretreatment inflammation and/or clinical depression enhance the odds of demonstrating omega-3 fatty acid-related improvements. The IFN-α trial clearly identified important benefits of omega-3 fatty acid treatment (
42).
Exercise
Considerable evidence supports the value of exercise in treating depression and preventing its onset (
76,
114,
115). Physically active individuals have lower levels of inflammatory biomarkers than their sedentary counterparts (
116); reductions in inflammation provide one potential explanation for the antidepressant benefits of exercise (
117).
In the Treatment With Exercise Augmentation for Depression (TREAD) study, patients with major depression who did not achieve remission following an adequate trial of a single selective serotonin reuptake inhibitor (SSRI) were randomized to two exercise augmentation groups (
118). The higher-dose exercise augmentation group had a 28.3% remission rate, compared with 15.5% for the lower-dose group, and effect sizes were the same or larger than those observed in pharmacological treatment augmentation studies (
118). Although four cytokines did not change significantly during the 12-week intervention, higher preintervention TNF-α levels were associated with larger decreases in depressive symptoms, and changes in IL-1β were correlated with changes in depressive symptoms (
119).
These TREAD study data are consistent with a paradox in the exercise literature. Despite the fact that observational studies reliably show that more active people have lower inflammation than their sedentary counterparts (
117,
120), data from randomized controlled trials demonstrating that exercise training reduces inflammation are sparse and inconsistent (
121,
122). In fact, two reviews of randomized controlled trials concluded that exercise produces little or no change in inflammatory markers in healthy people who do not lose weight (
121,
122).
However, just as higher pretreatment inflammation predicted a better response to a TNF-α blocker (
17), the TREAD study data suggest that exercise’s antidepressant effects may be greater in those who have higher pretreatment inflammation (
119). Similarly, the IFN-α trial (
42) demonstrated that treatment with omega-3 fatty acids was efficacious when individuals faced a major inflammatory challenge. In each case, the initial inflammatory profile made a difference.
Obesity
Depression promotes obesity, and obesity in turn promotes depression (
1,
123). Depressed people have a 58% higher risk of becoming obese; the risk for developing depression over time is 55% for persons with obesity (
123). Longitudinal studies suggest that depressive symptoms promote the development of the metabolic syndrome, which has central obesity as its cornerstone (
124,
125).
Depression and obesity have key inflammatory mechanisms in common. Obesity has been characterized as a state of chronic inflammation due to elevated plasma IL-6, TNF-α, and CRP levels (
1). What is more, the pathways are bidirectional; visceral adipose tissue’s secretion of proinflammatory cytokines can function as a stimulus for HPA axis activation, such that hypercortisolemia enhances adipocyte accumulation, and vice versa (
124).
Adiposity appears to fuel inflammatory stress responses. Women with greater central adiposity produced larger inflammatory responses to a laboratory stress task than their leaner counterparts (
126). Other authors reported that both higher BMI and greater body fat were associated with larger stress-induced IL-6 responses that did not habituate with repeated stress (
127).
Interactive Influence of Obesity and Age in Mice
A recent mouse study highlighted the joint impact of obesity and age on inflammatory responsiveness. Because both the decrease in lean body mass and the increase in adiposity that occurs with advancing years play a role in the age-associated increases in inflammation (
128), it was not surprising that immunotherapy induced a lethal cytokine storm in aged mice, but not young mice (
129). However, in young obese mice, immunotherapy induced the same cytokine overresponsiveness, organ pathology, and mortality as seen in the aged mice (
129).
Together, these convergent lines of evidence show how stress and depression can act synergistically with obesity to potentiate larger inflammatory responses that could in turn further fuel depression. In addition to higher baseline levels of inflammation, these data suggest that obesity also confers risk by generating larger inflammatory responses to stress or pathogens. Higher baseline inflammation provides an important substrate for subsequent exaggerated inflammatory responses to challenge.
Priming: Cross-Sensitization Among Cytokines, Stressors, and Depression
In addition to early life stress, other major life stressors function as proximal risk factors for major depression (
2). Both currently and formerly depressed people experience more major and minor stressors than those who do not have a depression history, and current and past depression can also boost emotional reactivity to stressors (
130–
132). Furthermore, depression can damage close personal relationships, a key stress buffer; both current and formerly depressed men and women had poorer family functioning than those who had no depression history, even years after their depression had remitted (
133). A history of depression may indicate a high-risk phenotype for stress responsiveness (
134). Accordingly, a past or current mood disorder could act synergistically with stress to heighten inflammation.
Cross-Sensitization in Rodents and Monkeys
The close tie between depression and stress has implications for inflammation; cross-sensitization between stressors and cytokines has been well documented in rats (
135,
136). For example, exposure to a novel environment, foot or tail shock, or even exposure to conditioned stimuli that were present during foot shock all served to enhance IL-6 production (
70,
135). Furthermore, rats that had previously been stressed produced larger and more rapid proinflammatory responses to a bacterial endotoxin than did rats without a prior stress exposure (
135).
The rats’ endotoxin exposure mimics the immune challenges that occur frequently in daily life. For example, high-fat meals can provoke mild postprandial endoxemia (
137,
138) as well as alterations in gut microbiota and intestinal permeability (
139).
These data are important because the physiological systems that respond to endotoxin also respond to behavioral challenges, and this shared responsivity is particularly detrimental when endotoxin exposure occurs in proximity to psychological stress (
140). In rodents, when endotoxin was paired with stressors such as tail shock or restraint, it synergistically increased production of proinflammatory cytokines, exceeding the effect of endotoxin or the stressor alone (
141). The situational context also substantially affected behavioral and immunological responses to low-dose endotoxin in rhesus monkeys; the potency of the stressor influenced the magnitude and nature of endotoxin responses (
140).
Recent mouse studies provide provocative evidence that higher IL-6 production may influence behavioral responses to social stress (
48). Both in vivo and in vitro IL-6 responses predicted subsequent behavioral responses to repeated social defeat. Mice with larger IL-6 responses to initial aggressor exposure later displayed a stress-susceptible behavioral phenotype (depressive-like behavior) and more persistent stress-related IL-6 elevations (
48). Those with smaller initial IL-6 responses were more likely to display subsequent resilient or dominant behaviors. There were also preexisting immune differences in stimulated IL-6 production between mice that would later display stress-susceptible versus resilient behavioral profiles. In further studies, a pharmacological IL-6 blockade prevented the development of social avoidance behavior, highlighting its key role (
48).
Accordingly, preexisting individual differences in IL-6 responsivity predict stress vulnerability (
48). Importantly, because these differences occurred within an inbred, genetically similar strain, both epigenetic and environmental factors (e.g., parental transmission of stress sensitivity, differences in the stability of the home cage’s social hierarchy, or postnatal microbial exposures) likely played a prominent role in developing these profiles (
142–
144).
Depression Primes Inflammatory Responsiveness
In accord with the animal literature, human studies show that depression primes inflammatory responses, promoting larger cytokine increases in reaction to stressors and pathogens. For example, mild depressive symptoms were associated with amplified and prolonged inflammatory responses following influenza vaccination in older adults as well as pregnant women (
145,
146). Among women who had just given birth, those who had a lifetime history of major depression showed greater increases in both serum IL-6 and the soluble IL-6 receptor after delivery than women without a depression history (
147). Similarly, patients with major depression had larger increases in inflammatory markers than nondepressed controls in response to a laboratory stressor (
148,
149). In another study, individuals with more depressive symptoms had larger stress-induced increases in IL-6 following laboratory stressors than those with fewer depressive symptoms (
150).
Studies that have addressed the impact of repeated laboratory stressors on IL-6 production do not show evidence of habituation (
127,
151). Thus, if both currently and formerly depressed people experience more stressors than those without a depression history (
130–
132), they would likely continue to experience repeated exaggerated inflammatory responses.
Just as individual differences in IL-6 responsivity predict stress vulnerability in mice, the IL-6 overresponsiveness in people with depression, childhood adversity, and obesity also reflects risk. Larger, more frequent, or more prolonged inflammatory responses have negative mental and physical health consequences.
Assessing Inflammation in Research and Clinical Practice
Inflammation signals responsiveness to certain therapies and provides a guide to potential targets for clinical symptom management. For example, a meta-analysis revealed that nonsteroidal anti-inflammatory drugs reduced depressive symptoms compared with placebo, particularly the selective COX-2 inhibitor celecoxib; patients with higher inflammation benefited most (
152). Similarly, a clear theme across research with omega-3 fatty acids, exercise, and cytokine antagonists is that anti-inflammatory interventions have a substantially greater impact on mood in individuals with heightened inflammation. Higher CRP levels were associated with a better response to escitalopram than to nortriptyline (
153). The antidepressant effects of anti-inflammatories may be magnified in patients with comorbid pain-related or inflammatory disorders—which cover a wide spectrum, from psoriasis to cardiovascular disease to obesity. Furthermore, individuals who have relevant inflammatory genetic polymorphisms or gene expression profiles may be more responsive to these treatments (
38,
39,
46,
49,
109,
154). It follows that substantial inflammatory changes (and benefits) may not be observed in those with lower inflammation who undergo the same treatment.
Indeed, a finding that highlights the importance of heightened inflammation in the context of treatment choice is that patients with lower inflammation who received a placebo improved more than did patients assigned to active treatment in both infliximab and omega-3 fatty acid trials (
17,
18). These findings led to the suggestion that anti-inflammatory therapy may be harmful for patients without inflammation-driven major depression (
17,
18).
Identification of patients who can benefit from anti-inflammatory interventions is clearly important.
Table 1 lists risk factors for heightened inflammation, an indirect way to evaluate a patient’s inflammatory phenotype.
For objective confirmation, the optimal strategy would be to determine treatment based on a set of biomarkers, with IL-6, TNF-α, and CRP having the strongest relationships with depression (
3,
4). Differences in inflammatory patterns are likely, given the heterogeneity of the major depression population (
173), and thus multiple biomarkers could provide a clearer picture of inflammatory status than a single assay, facilitating identification of the best candidates for anti-inflammatory interventions (
18). For example, in an omega-3 fatty acid trial, patients with high values on any one of five biomarkers were more responsive to EPA than to placebo, and the EPA-placebo differences were larger in those who had multiple heightened inflammatory markers (
18). After identification of anti-inflammatory treatment candidates, additional inflammatory assessments can provide useful treatment response data.
For routine clinical use, a biomarker needs to provide accurate and reproducible data with well-validated norms (
174). Hospital laboratories need standardized assays that provide replicable data across sites (
174). CRP meets these criteria, but none of the cytokines do, limiting their utility for clinical practice at present, despite their obvious value (
Table 2).
Anti-inflammatory treatment trials should preselect patients with heightened inflammation, and routinely assess inflammatory change (
175). Surprisingly, in anti-inflammatory depression trials to date, heightened inflammation has not been part of the inclusion criteria (
175). Using baseline inflammation to predict treatment response has provided provocative data, substantially expanding our understanding of who may benefit (
17,
18), a worthwhile approach for future studies. In addition, researchers should examine the extent of inflammatory change and relate it to changes in depressive symptoms to better understand how lowering inflammation influences depression.
Inflammation’s Implications for Treatment of Major Depression
Inflammation is not ubiquitous among people with depression, but when the two conditions co-occur, treating inflammation in tandem with depression can enhance recovery and reduce the risk of recurrence.
Table 3 includes anti-inflammatory pharmacological treatments for depression. A meta-analysis of the effects of antidepressant medication on cytokines showed an average 50% reduction in depressive symptoms across antidepressants, but only SSRIs appeared to affect cytokine production (
191).
Other interventions can also be beneficial. Alcohol dependence and smoking are often comorbid with depression, and both have notable inflammatory consequences (
169,
192); successful treatment of either can produce durable positive changes in both inflammation and depression. Lifestyle interventions (
Table 3), including weight loss, dietary change, exercise, and some integrative medicine interventions, can provide significant positive long-term benefits for patients who make and maintain a commitment to them.
Early interventions may be particularly important as a prophylaxis for those with predispositions to depression and heightened inflammation. For example, in a randomized study, inflammation was lower in 17-year-olds who had been assigned to a brief family intervention 8 years earlier compared with controls (
187).
The efficacy of cognitive-behavioral therapy (CBT) in depression treatment is well documented, and CBT may concurrently reduce inflammation (
55,
188). In fact, one nonrandomized trial found posttreatment decreases in an indicator of intestinal bacterial translocation as well as other inflammatory markers (
55).
Both pain and disturbed sleep boost inflammation (
69,
70,
75). Improvements in pain and sleep can enhance treatment outcomes and reduce the risk for recurrent depression (
73–
75). CBT has well-documented efficacy in the long-term remission of insomnia, and the addition of CBT for insomnia to a standard antidepressant regimen can produce a more rapid and longer-lasting remission than antidepressant treatment by itself (
193). Moreover, CBT for sleep disturbances can also reduce inflammation as well as depressive symptoms (
185,
189). For example, CBT for insomnia produced greater positive change (improvements in sleep, daytime fatigue, depressive symptoms, and CRP level) than either tai chi or a sleep education control condition, and remission of insomnia was associated with lower CRP levels 16 months after treatment (
185).
CBT for pain and pain-related problems has improved pain, physical disability, and mood across a range of chronic pain syndromes (
194). In addition, a randomized study with rheumatoid arthritis patients found that decrements in IL-6 levels were larger with CBT than with mindfulness meditation or an education-only condition (
190).
We have highlighted the impact of overresponsiveness to daily stressors as an important pathway deserving of greater attention. Indeed, exaggerated stress-induced inflammatory responses mark a number of conditions that increase depression risk—e.g., fatigue, loneliness, lower subjective social status, smoking history, and marital discord (
195–
199). Accordingly, exaggerated inflammatory responses could reflect a greater risk for depression. Inflammatory overresponsiveness may stem in part from decreased glucocorticoid stress responses (
26,
154,
195), and blunted glucocorticoid signaling in concert with increased NF-κB signaling may provide one functional fingerprint for chronic stress (
154). Consequently, interventions targeting overresponsivity may benefit mood and inflammation. For example, cognitive-behavioral treatments that mute affective overresponsiveness to stressors could have important protective effects. In addition, some evidence suggests that meditation and yoga may reduce inflammatory responsiveness (
181,
183,
184).
We have focused on how depression and inflammation are intertwined, but the implications extend to other health outcomes. Depression and inflammation are both linked to a number of disorders and systemic diseases, and the processes we described clearly have an impact on those diseases as well. Depression has a substantial global disease burden, and excess depression-related mortality has been documented in multiple diseases (
200). The bidirectional links between depression, inflammation, and disease make this research complex; they also suggest that effective depression treatments can have a far-reaching impact on mood, inflammation, and health.