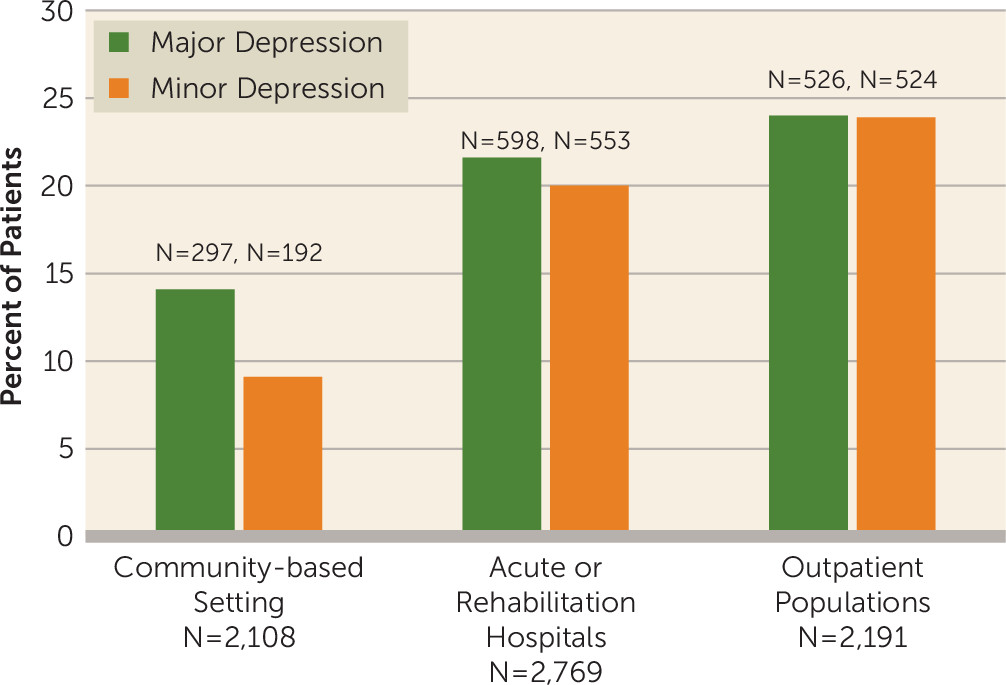Our keywords included “poststroke depression,” “depression AND cerebrovascular disorders,” “vascular depression,” “stroke AND antidepressant,” “stroke AND antidepressant AND depression,” “poststroke depression AND randomized clinical trial,” as well as “poststroke depression AND trial.”
Reference lists of each article utilized were searched by hand to identify additional citations not identified by the databases. This is primarily a narrative review that did not utilize the Preferred Reporting Items for Systematic Reviews and Meta-Analysis [PRISMA] because of the limitations of existing meta-analyses in this field discussed under the section on incidence and prevalence of PSD.
Diagnosis of PSD
The DSM-5 defines poststroke mood disorders as mood disorders due to stroke with depressive features, major depressive-like episode, or mixed-mood features. The only disorder in DSM-5 that is specific for cerebrovascular disease is major or minor vascular neurocognitive disorder.
A patient with a diagnosis of mood disorder due to stroke with a major depression-like episode must have depressed mood or loss of interest or pleasure along with four other symptoms of depression lasting 2 or more weeks. Patients with a diagnosis of mood disorder due to stroke with depressive features must have depressed mood or loss of interest or pleasure along with at least two but less than five symptoms of major depression lasting 2 weeks or longer.
Clinically defined vascular depression was proposed in 1997 by Alexopoulos et al. (
7), while Krishnan et al. (
8) validated such diagnosis on the basis of the presence of subcortical pathology and white matter hyperintensities in MRI scans.
Patients with vascular depression have later age at onset, greater cognitive impairment, less family and personal history of depression, and greater physical impairment than geriatric patients with nonvascular depression. In addition, patients with vascular depression with executive dysfunction and/or patients who show progression of white matter hyperintensities over time have a poor response to treatment with antidepressants and a more chronic and relapsing clinical course (
9).
Although vascular depression is related to small-vessel ischemia and PSD usually is related to large-vessel infarction (except lacunar infarcts), most studies have found similarities between these conditions, such as lower frequency of family and personal history of depression, prominent executive dysfunction, and greater disability compared with nonmicrovascular geriatric depression (
7,
10–
12). One study reported that the significant association between the presence of cardiovascular risk factors and the severity of depression observed in vascular depression is not present in PSD (
13). The existing evidence, however, suggests that PSD is one form of vascular depression. Thus, a single cerebral infarct may trigger the same pathophysiological changes of depression as slowly evolving vascular ischemia.
Incidence and Prevalence of PSD
The frequency of PSD has been studied in many countries of the world. The most-quoted studies of prevalence and incidence of PSD have utilized meta-analysis to create large databases (
14,
15). The most recent meta-analysis of 61 cohorts including 25,488 patients (
15) reported that 31% of patients developed depression at any time point up to 5 years following stroke. A prior meta-analysis of 43 studies published in 2013 included 20,293 patients and reported that the pooled prevalence of PSD was 29% at any time point within 5 years following stroke (
14). In addition, the investigators found that the cumulative percent of patients who developed one or more depressions within the first 5 years following stroke ranged from 39% to 52% (
14).
The contributions by Ayerbe et al. (
14) and Hackett and Pickles (
15) are significant because they 1) establish that poststroke depression is a frequent and important complication of stroke and 2) refute prior assertions that the frequency of PSD has been exaggerated (
16). However, these meta-analyses have included many studies that have defined PSD based on arbitrary cut-off scores on a depression rating scale. These scales provide information about the frequency and severity of depressive symptoms, but their use as a diagnostic instrument has rarely been validated. On the other hand, it has been clearly established that the existence of depression should be ascertained based on a structured mental state examination and should meet established diagnostic criteria for a specific depressive disorder (
17). Thus, these meta-analyses did not distinguish major depression from other forms of depressive disorders occurring after stroke, and many did not examine the time since stroke, the clinical setting (e.g., community or hospitalized patients), or the severity of stroke, all of which may affect the prevalence of depression. We conducted a pooled analysis of studies published prior to 2003 (
18) using only studies that used structured interviews and diagnostic criteria to identify major and minor depression in community-based settings, acute or rehabilitation hospitals, or outpatient clinics. The results are shown in
Figure 1 and demonstrate that the frequency of PSD depends on the clinical settings where the patients are seen, which is also related to the severity of stroke. Thus, our knowledge of important clinical differences in the prevalence and incidence of PSD has been lost in the existing meta-analyses of aggregated data.
The Impact of PSD on Stroke Recovery and Mortality and the Effects of Antidepressants
Numerous studies have examined the relationship between depression at the initial examination (which may range from a few weeks following stroke to 6 or more months following stroke) and functional and motor recovery (
36–
38). Five of six studies that examined whether severity of depression after acute stroke predicted severity of impairment in activities of daily living at 1 year or more of follow-up found that depression severity was an independent predictor of severity of impairment in activities of daily living (
18).
Consistent with the previous findings, patients with PSD who responded to treatment with nortriptyline or fluoxetine showed significantly better improvement in activities of daily living than patients with PSD who did not respond to active treatment or placebo (
39). Similarly, a longitudinal study has shown that response to treatment of PSD with nortriptyline or fluoxetine over 12 weeks leads to improved cognitive function to the level seen in nondepressed stroke patients that lasts for more than 2 years (
40).
Increased mortality associated with PSD is perhaps the most dramatic clinical phenomenon following PSD. The first study to report this phenomenon using standardized interviews to diagnose PSD was published in 1993 (
41). In this study, 103 patients were followed up 10 years after their index stroke to determine mortality rates. Patients who developed PSD during the acute poststroke period had significantly higher mortality rates than similarly impaired stroke patients with no in-hospital depression (odds ratio=3.4, 95% CI=1.4–8.4, p=0.007) (
41). A similar finding was reported by House et al. (
42), who showed that even mild severity of PSD was associated with increased mortality as early as 1 year after stroke. Furthermore, in a cohort of 51,119 veterans hospitalized because of an ischemic stroke, those who developed PSD had a higher 3-year mortality risk than veterans without a mental health diagnosis (
43).
A recent study of 1,354 patients that used the Hospital Anxiety and Depression Scale to assess PSD found that at the 5-year follow-up, patients with a Hospital Anxiety and Depression Scale score ≥7 at 3 months after stroke had a hazard ratio of 1.41 (95% CI=1.13–1.77, p=0.02) of increased mortality compared with patients with scores <7 at 3 months (
44,
45). The investigators also reported increased mortality among patients started on selective serotonin reuptake inhibitor (SSRI) antidepressants in the 3 months after stroke. However, this was not a randomized trial, and the data analysis on which this assertion is based does not include all the relevant confounders such as disability, depression severity, and the effect of comorbid medical conditions (
44).
The association of PSD with mortality appears to be the result of an increase in cardiovascular mortality (
44).We reported that decreased heart rate variability, which has been demonstrated to play an etiological role in mortality associated with depression and myocardial infarction (
46), was also associated with PSD (
47). Thus, disruption of autonomic system function in patients with PSD might contribute to cardiovascular mortality.
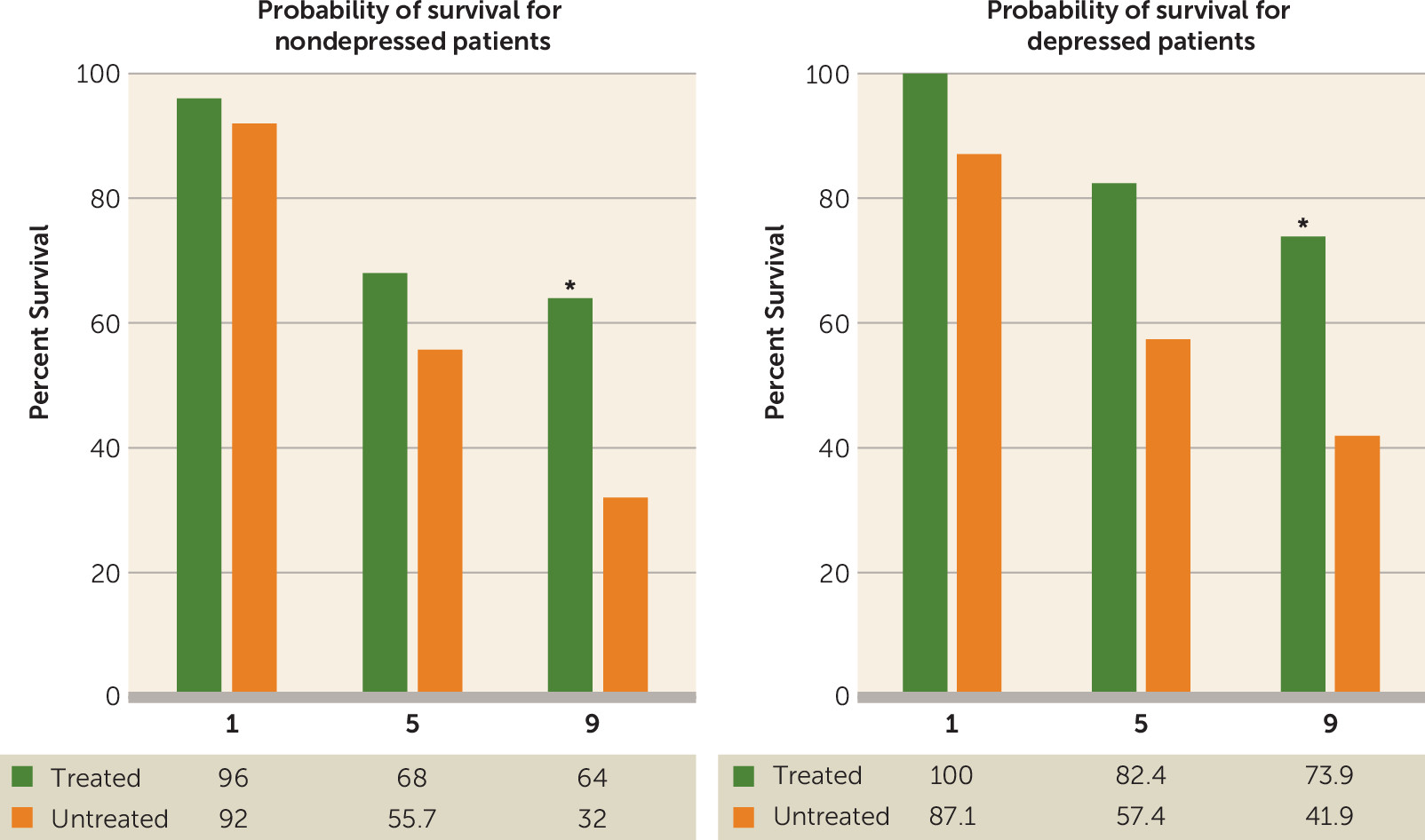
Perhaps the most provocative finding, however, was the relationship of mortality following PSD to treatment with antidepressants (
Figure 2) (
48). A 9-year follow-up study of patients with or without PSD who had been treated with nortriptyline (100 mg/day) or fluoxetine (40 mg/day) for 12 weeks (N=53) showed that patients receiving active treatment had an increased probability of survival at the 9-year follow-up compared with similar patients given placebo (N=28) (i.e., 59.2% survival for patients treated with nortriptyline or fluoxetine compared with 34.6% for patients given a placebo [adjusted odds ratio=3.7, 95% CI=1.1–12.2, p=0.03]) (
48). This finding held when nortriptyline- and fluoxetine-related survival was examined separately and was independent of whether the depression responded to treatment or whether the patient was depressed or not prior to treatment. A beneficial effect of antidepressants on long-term survival after stroke was also observed in a group of 790 veterans with stroke followed over a 7-year period (
49).
Three studies analyzed the effect of antidepressants on motor, cognitive, and disability outcomes among nondepressed patients during 12 months following stroke. Chollet et al. (
50) found that 12 weeks of fluoxetine (20 mg/day) was more efficacious than placebo to improve motor recovery at 12 weeks assessed using the Fugl-Meyer Motor Disability Scale. Jorge et al. (
51) reported that among a group of nondepressed patients with acute stroke, 12 months of escitalopram (5 mg–10 mg/day) enhanced cognitive performance at 12 months assessed by the Repeatable Battery for the Assessment of Neuropsychological Status.
With regard to disability, a secondary analysis of the results of our previous randomized controlled trial on the efficacy of fluoxetine (20 mg–40 mg/day) and nortriptyline (100 mg/day) to treat PSD showed that when compared with those receiving placebo, nondepressed stroke patients receiving antidepressants had decreased disability measured by the modified Rankin Scale at the 12-month follow-up (
52).
In summary, although two of the factors most consistently associated with PSD are functional disability and cognitive impairment, there is clearly a reciprocal relationship with impairment influencing depression and depression influencing impairment. In addition, there is rapidly growing literature indicating that SSRIs following stroke may enhance recovery independent of PSD or their effects on mood (
50–
52). The findings of a recent meta-analysis of studies assessing the effect of SSRIs on stroke outcomes support this conclusion (
53).
Finally, a multicenter randomized controlled trial of the efficacy of citalopram to reduce disability and cardiovascular mortality following stroke (The Efficacy of Citalopram Treatment in Acute Stroke [TALOS] study) is currently underway (
54).
Etiological Mechanisms
Like many disorders in psychiatry, there is evidence supporting the role of psychological, social, and biological factors in the mechanism of PSD (
55). The most consistent finding in the stroke literature is that PSD is associated with stroke severity and the degree of functional physical and cognitive impairment (
23). However, it is uncertain whether the level of impairment is etiologically associated with the development of PSD through a “reactive” psychological mechanism or whether there are biological factors related to brain damage that contribute to the bidirectional relationship between disability and depression.
The association between PSD and biological factors has included empirical evidence suggesting alterations in ascending monoamine systems (
56), hypothalamic-pituitary-adrenal (HPA) axis abnormalities (
57), disruption of prefrontal-subcortical circuits (
58), alterations in neuroplasticity and in glutamate neurotransmission (
59), and an excess of proinflammatory cytokines (
60). However, a pathophysiological hypothesis of PSD that can integrate these changes into a coherent explanatory model has yet to be formulated.
From a translational approach, there have been several attempts to model PSD in experiments with rodents. We conducted the earliest studies using a rat model of middle cerebral artery ligation. Right hemisphere middle cerebral artery ligation produced significant bilateral depletions of norepinephrine and dopamine in both the cortex and brainstem, as well as hyperactivity and other behavioral changes not seen in rats given sham ligations (
61). Further experiments using this animal model demonstrated that the biogenic amine and behavioral effects were lateralized, not occurring after left middle cerebral artery ligation, and lasted up to 4 weeks (
62).
In recent years, there has been a renewed interest in modeling the neuropsychiatric consequences of stroke. Kronenberg et al. (
63) used a model of mild stroke in mice (i.e., 30-minute middle cerebral artery occlusion that limits the ischemic damage to the basal ganglia). A subgroup of these mice was given citalopram, starting 1 week after middle cerebral artery occlusion. Left, but not right, middle cerebral artery occlusion produced delayed degeneration of dopaminergic neurons in the left ventral tegmental area, which resulted in reduced dopamine concentrations in the striatum. The behavioral correlates of these neurodegenerative and neurochemical changes were anhedonia and behavioral despair assessed by the sucrose consumption test and forced swimming test, respectively. Chronic citalopram treatment initiated 7 days after stroke, however, prevented the degeneration of dopaminergic neurons and reversed the behavioral phenotype (
63).
There have also been animal studies that combined experimental ischemic lesions with social isolation or chronic mild stress protocols implemented during the recovery process after stroke (
64). Permanent left middle cerebral artery occlusion with a 14-day chronic mild stress protocol produced a depressive phenotype characterized by decreased exploratory behavior and sucrose consumption, a behavioral effect that was reversed by the administration of citalopram and a 5-HT
1A antagonist along with evidence of increased neurogenesis in the hippocampus (
64).
Unfortunately, there have been relatively few studies of the mechanisms of PSD in clinical populations. Decreased CSF levels of serotonin or norepinephrine metabolites, however, were significantly associated with severity of PSD (
65). It has also been hypothesized that ischemic lesions of ascending monoamine pathways may result in depressive disorders due to abnormal modulation of frontal and cingulate regions involved in mood regulation (
18).
There is a growing consensus that ischemic lesions (single and/or multiple) of the neural circuits that connect the prefrontal cortex, basal ganglia, thalamus, and amygdala (independently of their lateralization) may disrupt mood regulation and executive function leading to a similar clinical presentation. Furthermore, there appears to be a threshold by which the confluence of multiple etiological factors or further damage to specific white matter tracts, such as the cingulate bundle, the uncinate fasciculus, and superior longitudinal fasciculus, triggers the onset of clinical depression. Thus, acute ischemia can unveil the presence of vascular depression (
58), or a strategically located stroke might produce depression independently of the presence of widespread cerebrovascular pathology.
From a network standpoint, resting connectivity patterns revealed by functional MRI appear to be similar in vascular depression and PSD, with increased activation of the default mode network and limbic structures and decreased activation of task-related networks and the dorsolateral aspects of the prefrontal cortex (
66).
Earlier studies also examined the occurrence of HPA axis deregulation in PSD, particularly the association of PSD with elevated cortisol levels and abnormal negative feedback control of cortisol secretion (
57). More recently, a number of studies have provided support for the role of proinflammatory cytokines in the development of PSD (
56,
60). This hypothesis has been supported by recently published studies in which increased serum concentrations of interleukin-6 (Il-6) were associated with increased severity of somatic symptoms of PSD (
67). There is a bidirectional relationship between HPA axis deregulation and the levels of inflammatory cytokines by which high cortisol levels induce an inflammatory response that, in turn, will result in further HPA axis deregulation. Furthermore, increased levels of inflammatory cytokines reduce the synthesis and availability of serotonin through their enhancing effect on the activity of the enzyme indolamine 2, 3-dioxygenase (
56).
The recent emphasis on neuroplasticity as a critical neurobiological substrate for depressive disorders suggests that synaptic alterations in the prefrontal cortex and hippocampus may be etiologically implicated in PSD. In support, BDNF levels, one of the regulators of these processes, have been shown to be reduced in PSD (
68). A recent study of 216 patients with acute ischemic stroke reported the results of a multivariate analysis showing that lower serum levels of BDNF at admission were an independent predictor of PSD at the 3-month follow-up (
69). Furthermore, a meta-analysis performed by Noonan et al. (
59) based on 33 studies reported that lower serum BDNF was found in patients with PSD compared with nondepressed control subjects. Additionally, HPA axis deregulation and increased levels of proinflammatory cytokines may inhibit neurogenesis in the hippocampus and decrease the neuroplasticity of the prefrontal cortex contributing to the onset and perpetuation of PSD (
70).
Finally, a preliminary study using magnetic resonance spectroscopy found altered glutamate levels in the anterior cingulate cortex of depressed stroke patients (
71).
Thus, although there are numerous possible physiological mechanisms related to PSD, many investigators have concluded that this complex disorder, like most of the major psychiatric disorders not associated with stroke, may best be described as a bio-psycho-social disorder. However, this general theoretical framework does little to help us elucidate pathophysiological mechanisms leading to specific symptoms. These different etiological factors described above may have more salient roles in some forms or symptoms of PSD, and also their effects may vary at different times after the stroke. We believe future studies should attempt to identify the mechanism of specific symptoms or clinical characteristics of PSD rather than the whole syndrome.