Phosphodiesterase 10A in Schizophrenia: A PET Study Using [11C]IMA107
Abstract
Objective:
Method:
Results:
Conclusions:
Method
Participants
Clinical Assessment
Image Acquisition
[11C]IMA107 PET Data Processing
Movement minimization and correction.
Data analysis.
Image processing and definition of regions of interest.
Kinetic modeling.
Statistical Analysis
Results
| Measure | Schizophrenia Patients (N=12) | Healthy Comparison Subjects (N=12) | ||
|---|---|---|---|---|
| N | % | N | % | |
| Male | 8 | 67 | 8 | 67 |
| Right-handed | 12 | 100 | 12 | 100 |
| Education | ||||
| High school diploma (GCSE) | 6 | 50 | 2 | 17 |
| High school diploma, advanced (A-level) | 3 | 25 | 4 | 33 |
| University | 3 | 25 | 6 | 50 |
| Antipsychotics used (% of atypicals) | 9 | 75 | ||
| Mean | SD | Mean | SD | |
| Age at scan (years) | 33.16 | 6.88 | 32.58 | 7.06 |
| Cotinine blood level (ng/ml) | 372.90 | 345.30 | 296.70 | 287.90 |
| Caffeine blood level (mg/L) | 3.10 | 0.60 | 3.08 | 0.23 |
| Positive and Negative Syndrome Scale score | 61.50 | 9.70 | ||
| Antipsychotic dosage (mg per day of chlorpromazine equivalents) | 259.70 | 169.90 | ||
| Radioactivity injected (MBq) | 275.30 | 43.50 | 261.20 | 57.90 |
| Specific activity (GBq/μmol) | 2.95 | 1.74 | 3.25 | 1.80 |
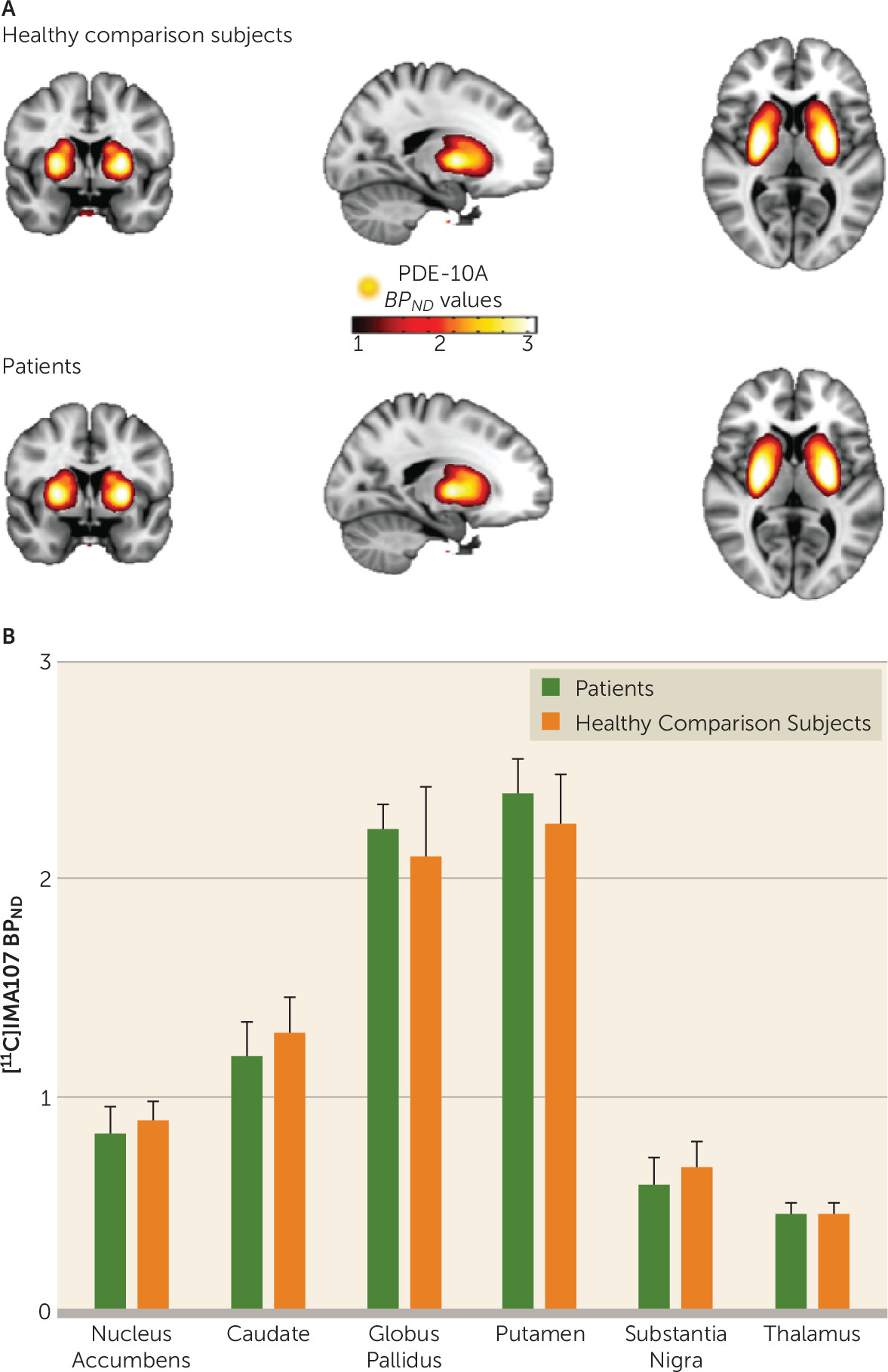
| Schizophrenia Patients (N=12) | Healthy Comparison Subjects (N=12) | % Change in Patientsa | |||
|---|---|---|---|---|---|
| Region of Interest | Mean | SD | Mean | SD | |
| Thalamus | 0.47 | 0.09 | 0.43 | 0.09 | 8 |
| Substantia nigra | 0.63 | 0.16 | 0.67 | 0.18 | –6 |
| Caudate | 1.26 | 0.20 | 1.28 | 0.26 | –1 |
| Putamen | 2.39 | 0.35 | 2.26 | 0.35 | 6 |
| Nucleus accumbens | 0.87 | 0.18 | 0.88 | 0.15 | –2 |
| Globus pallidus | 2.20 | 0.30 | 2.17 | 0.40 | 2 |
Correlations Between PDE-10A Availability and Clinical Characteristics
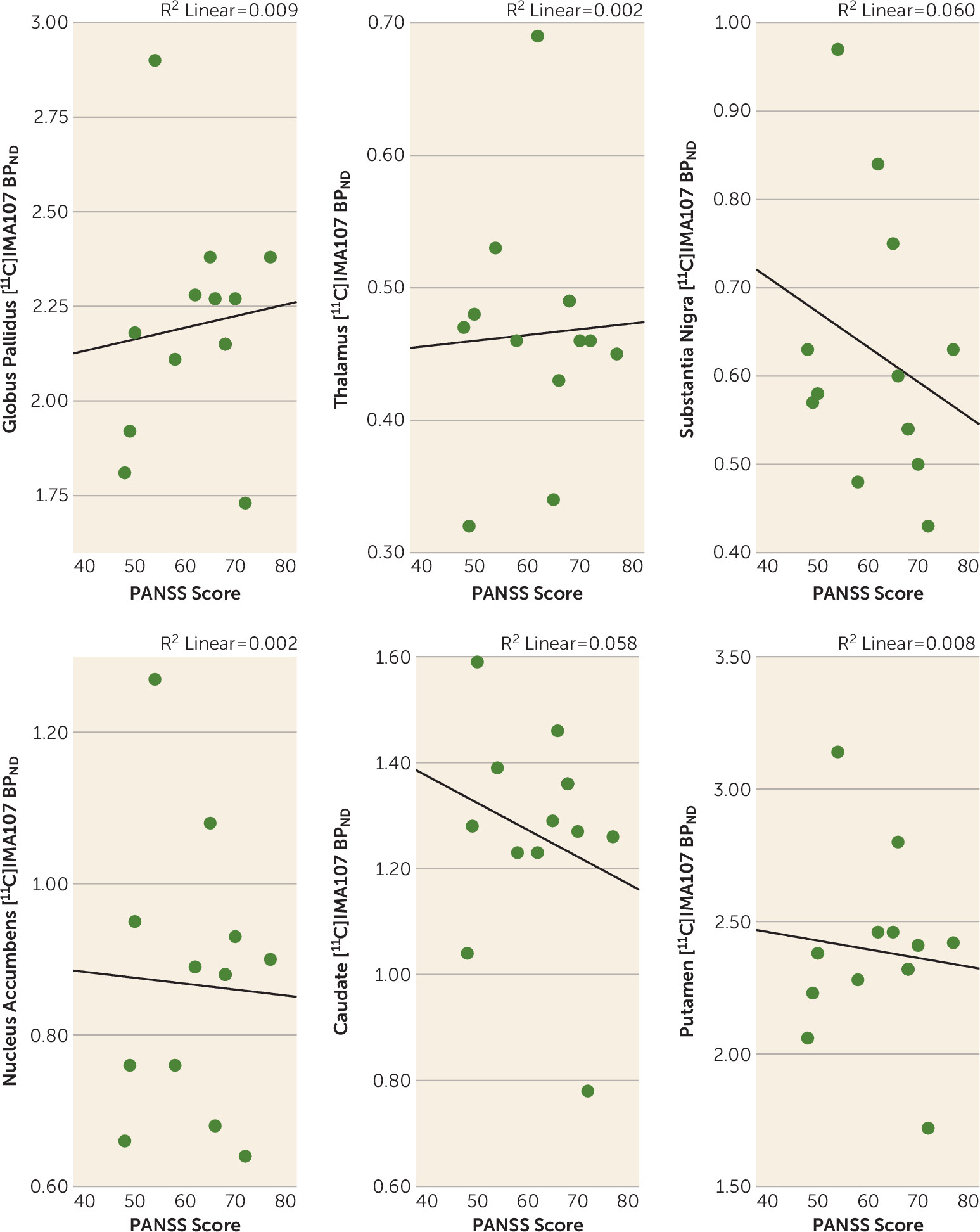
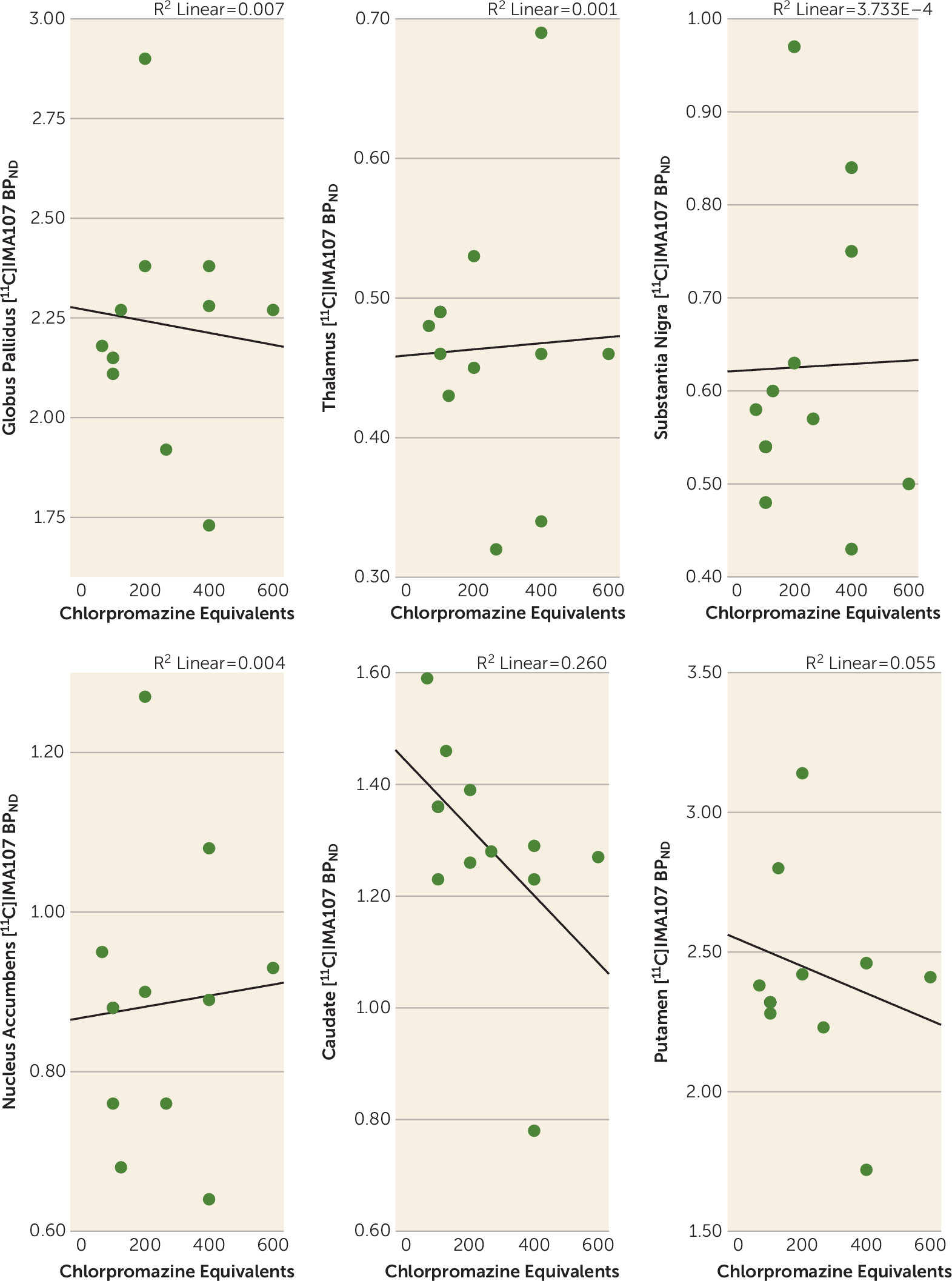
Discussion
Footnote
Supplementary Material
- View/Download
- 149.90 KB
References
Information & Authors
Information
Published In


History
Authors
Competing Interests
Funding Information
Metrics & Citations
Metrics
Citations
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
For more information or tips please see 'Downloading to a citation manager' in the Help menu.
View Options
View options
PDF/EPUB
View PDF/EPUBLogin options
Already a subscriber? Access your subscription through your login credentials or your institution for full access to this article.
Personal login Institutional Login Open Athens loginNot a subscriber?
PsychiatryOnline subscription options offer access to the DSM-5-TR® library, books, journals, CME, and patient resources. This all-in-one virtual library provides psychiatrists and mental health professionals with key resources for diagnosis, treatment, research, and professional development.
Need more help? PsychiatryOnline Customer Service may be reached by emailing [email protected] or by calling 800-368-5777 (in the U.S.) or 703-907-7322 (outside the U.S.).