Schizophrenia is a common psychiatric disorder defined by delusions, hallucinations, disorganized thought, social withdrawal, and various other symptoms that often emerge in early adulthood. The underlying molecular pathology likely includes broad changes in gene expression in neurons residing in the cerebral cortex and other brain regions, with many of the DNA polymorphisms implicated in heritable liability for schizophrenia positioned in gene proximal promoters and other “regulatory” DNA elements governing transcription (
1). A deeper understanding of the specific gene expression patterns that are dysregulated in the brain of individuals diagnosed with schizophrenia (and related disorders sharing some of the same genetic risk architectures and disease mechanisms, including bipolar disorder and some cases of unipolar depression) is critical. This is because even today, more than a half century after introduction of the first-generation antipsychotic drugs, at least one-third of the patient pool shows a poor treatment response to many of the conventional drugs, including those that target dopamine and serotonin signaling pathways (
2). Cognitive deficits, considered good predictors for long-term prognosis in schizophrenia (
3,
4), often are notoriously difficult to treat with our currently available psychopharmacological toolboxes. Therefore, the study of nonmonoamine-based neurotransmitter systems could provide important clues that ultimately could radically advance the existing treatment options for schizophrenia and psychosis. In this issue of the
Journal, a team led by Barbara K. Lipska, Ph.D. (
5) quantified, in the prefrontal cortex of approximately 380 case subjects diagnosed with schizophrenia and related disease and approximately 325 comparison subjects collected from fetal state to old age, the
CHRFAM7A and
CHRNA7 gene transcripts, encoding nicotinic acetylcholine receptor (nAchR) “α7” subunits. Note that the nAchRs are ligand-gated ion channels designed for fast millisecond-scale synaptic transmission, not to be confused with slower G protein-coupled muscarinic receptor systems targeted by benztropine and other anticholinergic drugs to treat or prevent extrapyramidal side effects from antipsychotics. This new study, on the backdrop of previous evidence from genetics, neuropathology, neurophysiology, and clinical pharmacology implicating nAchR in the pathophysiology of schizophrenia (
6,
7), offers important new insights, with particular relevance to the molecular mechanisms that could contribute to cortical dysfunction and diseased cognition.
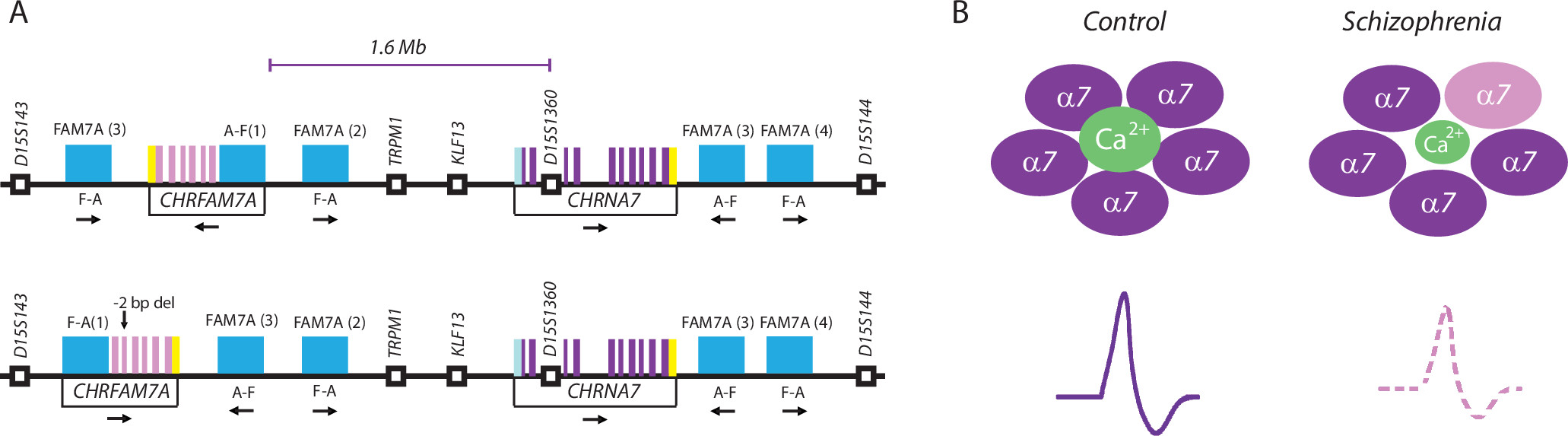
To provide some background, chromosome 15q13.3 is home to both genes that are center stage in this study,
CHRNA7 and
CHRFAM7A (Figure 1A). The latter gene is partially comprised by sequences highly homologous to
CHRNA7 exons 5–10 because of ancestral gene duplication events dating back to the time when the hominid lineage split from the rest of the primate tree (
7). Thus,
CHRFAM7A (which can be further dichotomized into two different variants in the human population [
Figure 1A]) is unique to humans and absent from the genome of the great apes and other mammals. Presently, little is known about
CHRFAM7A function. Interestingly, only a small fraction of
CHRFAM7A RNA appears to be translated into protein, in contrast to
CHRNA7, which contributes to a significant fraction of nAChR protein in the cerebral cortex and hippocampus (
7). But once synthesized inside the neurons, CHRFAM7A proteins are quickly coassembled with (
CHRNA7 encoded) α7 proteins in the cell membrane (including synaptic sites) (
8), which could effectively reduce signaling through α7 nAchR channels at pre- and postsynaptic sites of excitatory and inhibitory cortical synapses (
7).
The main findings of this study are that the ratio of
CHRFAM7A over
CHRNA7 transcript was significantly increased in subjects with schizophrenia or bipolar disorder, which at least in the schizophrenia cohort was associated with a significant deficit in
CHRNA7 transcript. Increased
CHRFAM7A/CHRNA7 ratios are also found in immature (including prenatal) prefrontal cortex in the absence of apparent psychiatric disease, leaving the authors to speculate about a neurodevelopmental origin of these alterations in the (adult) cortex of their clinical cases. As an alternative explanation, activity-dependent gene expression, in response to the prefrontal dysfunction that often affects patients with mood and psychosis spectrum disorders, could trigger secondary adaptations that mimic some of the transcriptional signatures that define “normal” immature cortex. Interestingly, neither smoking nor antipsychotic drug treatment history appeared to detectably affect
CHRNA7 and
CHRFAM7A RNA levels. This finding is of interest given that smoking reportedly induces robust increases in cortical nAchR receptor ligand binding (
7), which in light of the present findings then would point to posttranscriptional or even posttranslational mechanisms, rather than changes in gene expression, as the underlying cause.
What are the implications of this novel study, and how could this impact the field? Because the two transcripts,
CHRNA7 and
CHRFAM7A, are encoded not only within the same portion of chromosome but also, at least in the cerebral cortex, coexpressed by the same neurons, the authors speculate that the relative excess of
CHRFAM7A, as observed in some of their disease cohorts, could result in diminished synaptic signaling through the α7 nAchR, which in turn is likely to negatively affect cortical function and cognition (
Figure 1B). Given that α7 nAchRs (which are pentameric ion channels, assembled from five individual subunit proteins) harboring CHRFAM7A protein have fewer ligand binding sites (because CHRFAM7A protein, in contrast to CHRNA7 protein, lacks such sites), these new findings nicely fit with earlier reports on reduced α7 nAchR ligand binding in the cortex and hippocampus of some individuals with schizophrenia (
7). The new findings would also explain, in part, the high incidence of smoking in subjects with schizophrenia because smoking, as mentioned above, increases nAchR receptor ligand binding in the cortex (
7). Thus, smoking could be viewed as an adaptive response to increase cortical nAchR signaling, in order to counteract the molecular defects imposed by the imbalances in
CHRFAM7A and
CHRNA7 expression.
Clearly, given the tremendous importance to develop new treatments targeted toward the cognitive defects in schizophrenia, one would expect that the findings reported in this article will now jump-start the field and trigger a fresh wave of research on the nAchR as it pertains to cognition and schizophrenia. For example, while this study had for postmortem standards an impressive sample size (>700 brains), it essentially was confined to a single quantitative approach—relative abundance of RNA molecules measured by polymerase chain reaction—which does not allow solid conclusions on the true magnitude of the observed alterations on a case-by-case basis. It would be important, therefore, to develop additional tools, such as antibodies with highly specific affinity for the CHRFAM7A and CHRNA7 proteins (or either one). These and other protein-based approaches would allow further testing of the working hypotheses as put forward in this study. Equally important, it will be necessary to harness preclinical model systems (including genetically engineered animals expressing the human-specific
CHRFAM7A gene) to study brain and behavior in the context of α7 nAchR ion channels comprised of CHRFAM7A and CHRNA7 subunits. In addition, it may be interesting to comb through the “genomic neighborhood” of the critical two-megabase portion of chromosome 15q13.3 that houses both the
CHRFAM7A and
CHRNA7 genes (
7) (
Figure 1A) and identify sequence elements that could serve as, for example, transcription factor or small RNA (“microRNA”) targets, or chromosomal loop organizers (
9), and thereby affect the orderly balance of
CHRFAM7A and
CHRNA7 expression. Finally, it is important to further examine the aforementioned hypothesis that smoking, which upregulates α7 nAchR receptor protein and ligand binding (
7), is a compensatory response to overcome the deficits in α7 nAchR ligand binding and signaling as imposed by dysregulated expression of
CHRFAM7A and
CHRNA7. Intriguingly, another new study, also published in this issue (
10), provides compelling evidence that smoking prospectively predicts risk for schizophrenia. Thus, by connecting the dots (
5,
10), it is plausible to speculate that imbalanced expression of
CHRFAM7A/CHRNA7 could long precede the onset of overt clinical symptoms, with nicotine abuse and dependence as early warning signs of increased liability for future psychosis in susceptible individuals. Undoubtedly, the chromosomal neighborhood (chromosome 15q13.3), home to
CHRFAM7A and
CHRNA7 (
Figure 1A), needs to be watched closely!