The incidence of anxiety disorders peaks during adolescence (
1). More than 75% of adults with anxiety disorders met diagnostic criteria as children or adolescents (
2,
3). However, because of a lack of sufficient diagnoses or specialized therapeutics, less than one in five children or adolescents with anxiety will receive treatment (
4). In 2004, the U.S. Food and Drug Administration (FDA) issued a black-box warning for selective serotonin reuptake inhibitors (SSRIs), the main class of pharmacological agents used to treat depression and anxiety disorders, for children and adolescents because of a risk of suicidality (
5). Within 2 years after the FDA advisory was issued, SSRI prescription rates for pediatric populations decreased (
6,
7). However, the impact of SSRIs on brain development during adolescence remains unknown.
Preclinical studies in rodents and nonhuman primates have shown that administration of SSRIs during periadolescence leads to persistent neurochemical changes into adulthood. In both rodent and nonhuman primates, a prolonged up-regulation of the serotonin transporter (SERT) has been found in the cortex and the hippocampus after fluoxetine treatment during the juvenile or periadolescent stages of life (
8–
10). Interestingly, in primates, no effects were observed on fear-related or social behaviors (
8). In rodents, early-life fluoxetine was found not to change anxiety-like or fear extinction behaviors in adulthood (
11–
13), although conflicting reports exist (
14,
15). Notably, all preclinical studies were performed in wild-type animals that did not exhibit elevated depressive or anxiety-like phenotypes, for which SSRIs are indicated in human populations.
In this context, genetically engineered mice expressing reduced levels of brain-derived neurotrophic factor (BDNF), a neurotrophin involved in neuronal growth and plasticity, display increased anxiety-like and depressive-like behaviors (
16,
17). Additionally, SSRI-associated up-regulation of BDNF is thought to be a key mechanism by which the long-term effects of SSRIs on neuronal plasticity are mediated (
18,
19). This genetic knock-in mouse model of a common single-nucleotide polymorphism (SNP) in the human
BDNF gene (rs6265) may hold particular relevance to human populations, as this SNP in humans is associated with altered susceptibility to anxiety and depressive pathology (
20–
22). This SNP leads to substitution of the conserved valine with a methionine at position 66 in the BDNF polypeptide (
17), resulting in decreased BDNF bioavailability (
23,
24). Of note, BDNF Val66Met mice reproduce the phenotypic hallmarks of human carriers, including altered anxiety- and fear-related behaviors (
17,
25), and this phenotype is not responsive to fluoxetine administered in adulthood (
17,
26).
In this study, we sought to determine whether developmentally timed SSRI administration in BDNF
Met/Met mice during periadolescence would lead to persistent neurochemical and behavioral changes in adulthood. The periadolescent period, which corresponds to the transition from childhood to adolescence, is when BDNF levels rise significantly (
27,
28). Thus, we hypothesized that pharmacological intervention during periadolescence, which would further elevate both serotonin and BDNF levels, may alter subsequent developmental trajectories for the neuronal populations dependent on these neuromodulators and alter the emergence of anxiety and fear-related behavioral phenotypes in the BDNF
Met/Met mice.
Discussion
Anxiety disorders emerge in childhood and adolescence (
52–
55), a developmental time frame characterized by transitional changes in emotional behavior as well as cortico-limbic neurocircuit maturation (
56). Here, we identify in a genetic mouse model of the common human BDNF SNP a “sensitive period” during the transition into adolescence (P21–P42) in which a timed pharmacological intervention rescued subsequent expression of elevated anxiety-like phenotypes. We found that the anxiety-like behaviors in BDNF
Met/Met mice, which emerge in late adolescence (
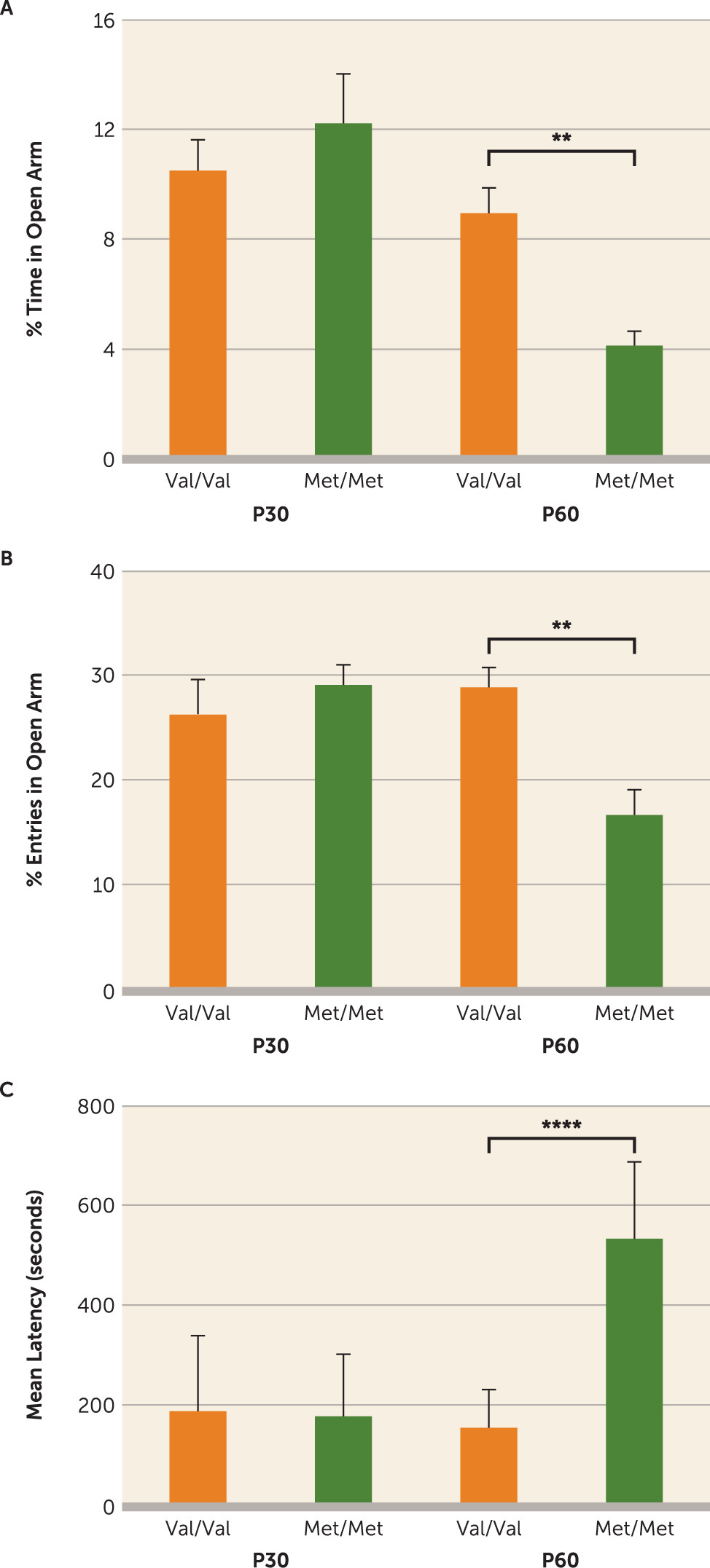
Figure 1) (
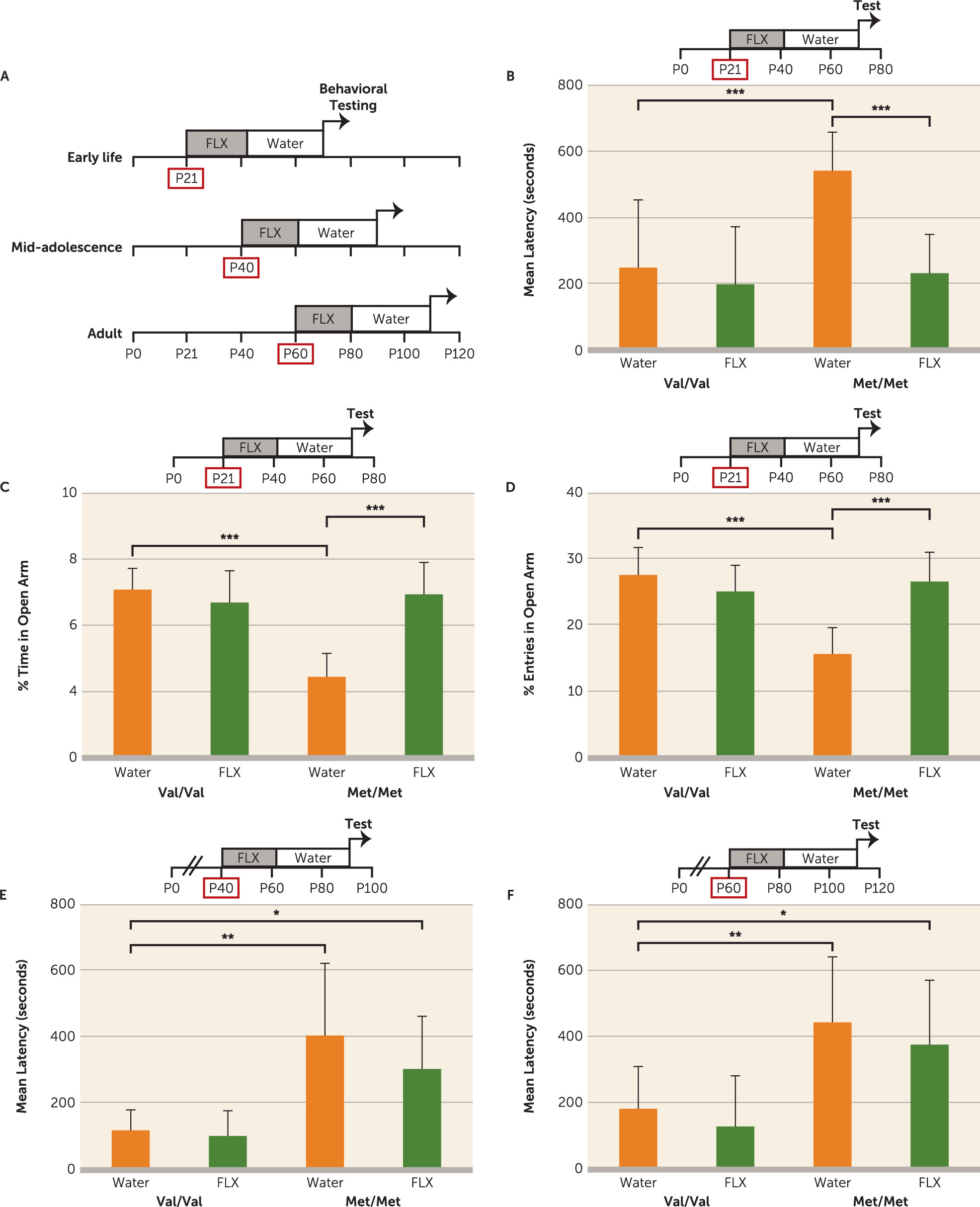
36), were rescued with early-life (P21–P42) fluoxetine treatment, which persistently elevated a serotonergic trophic factor, S100B, and enhanced maturation of serotonergic projections particularly to the infralimbic prefrontal cortex. To our knowledge, this is the first report of a developmentally timed pharmacological treatment that fully rescues a genetically induced anxiety-related behavioral phenotype during a discrete developmental window.
This P21–P42 “sensitive period” in BDNF
Met/Met mice is well outside the limits of a previously identified early postnatal window when environmental perturbation of serotonin signaling leads to persistently altered emotional behaviors. Postnatal (P4–P21) exposure to the serotonin inhibitors fluoxetine, clomipramine, or citalopram but not desipramine, a norepinephrine transporter inhibitor, has been shown to elevate anxiety-like behaviors in adult wild-type animals (
12,
57–
59). However, there are conflicting reports on whether SSRI exposure in periadolescence and adolescence has effects on adult emotional behavior. Mice receiving fluoxetine during the P14–P42 period exhibit an anxiogenic response when behavior is assessed immediately after treatment, although the effect is lost in adulthood (
13). In addition, adolescent (P12–P21, P21–P49, P22–P41) or adult (P56–P84) fluoxetine administration did not lead to long-term changes in anxiety-like or fear extinction behaviors in wild-type mice (
11,
12). On the other hand, rats receiving fluoxetine during the P25–P46, P35–P49, or P67–P88 periods spend significantly less time in the open arm of the elevated plus maze at 1–3 weeks after drug cessation (
14,
15,
60), which suggests species-specific effects of SERT blockade across development (
61). The reported discrepancies could be due to a number of methodological factors, such as the dosage, delivery route, and duration of fluoxetine treatment. Our study shows that wild-type mice do not exhibit long-term alterations in anxiety and fear learning behaviors when treated during the P21–P42 period. These findings are in accordance with previous studies showing that fluoxetine administration during adolescence, including the P21–P49 period, or during adulthood does not lead to long-term changes in anxiety-like or fear extinction behaviors in wild-type mice (
11,
12). Furthermore, it is important to point out that the anxiolytic fluoxetine effects we observed in BDNF
Met/Met mice are due to developmental compensation of trophic support.
Additionally, it has been shown that early-life exposure to fluoxetine or ketamine may have persistent antidepressant effects in rodents. Fluoxetine exposure at P35–P49 in mice resulted in suppressed depression-like behavior (
14). Repeated ketamine exposure in rats at P35–P49 resulted in anxiolytic- and antidepressant-like responses 2 months after drug exposure (
62). Furthermore, fluoxetine exposure during adolescence (P35–P49) in rats resulted in long-lasting decreases in behavioral reactivity to forced swimming stress (
60). These reports highlight the need for further research into the enduring impact antidepressants may have on the developing nervous system.
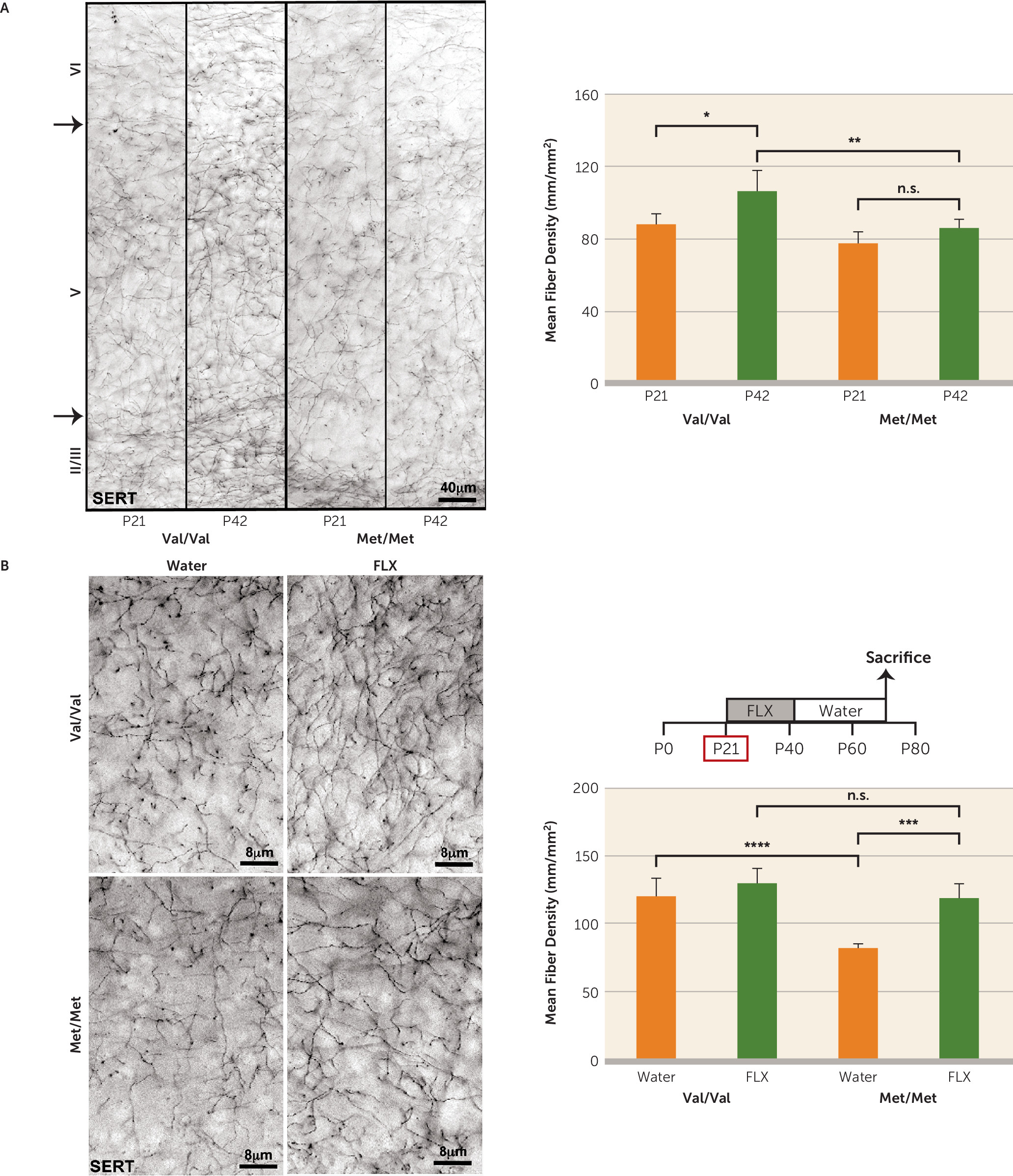
Previously, the variant BDNF Met allele has been associated with decreased 5-HT
1A receptor binding as well as SERT density in the cortex and hippocampus in human carriers (
44,
45). In accordance, BDNF
Met/Met mice exhibit perturbations in the serotonergic system as a result of reduced trophic support (
Figure 3A,B). Our results are also consistent with a report from a study in BDNF haploinsufficient (BDNF
+/−) mice indicating a reduction in serotonergic fibers in the cortex and hippocampus of these mice (
46).
Interestingly, our results indicate that early-life fluoxetine alters the density and complexity of serotonergic projections in BDNF
Met/Met mice (
Figure 3A,B), suggesting that at P21–P42 serotonergic neurons in BDNF
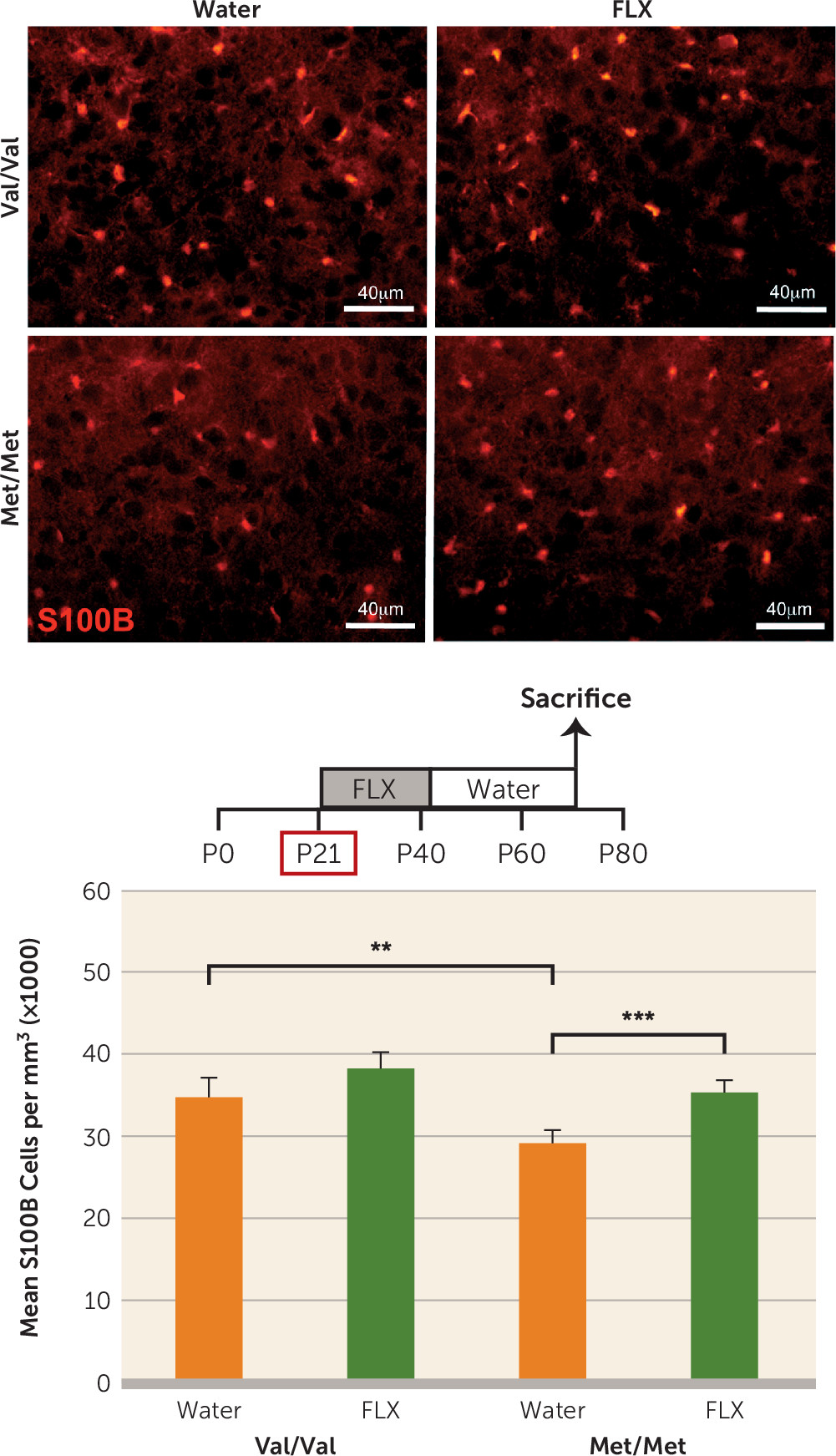
Met/Met mice, but not in wild-type mice, are still responsive to changes in environmental stimuli. Administration of fluoxetine to rats during periadolescence (P21–P35) results in significant increases in S100B in multiple brain regions that persist into adulthood (
63). In vitro and in vivo studies indicate that S100B has trophic effects on serotonergic neurons (
50,
64,
65). Indeed, 10-week-old mice overexpressing S100B exhibit increased density of serotonergic fibers in the hippocampus (
66). Persistent elevation in S100B expression induced by fluoxetine administered during periadolescence (
Figure 4) may mediate the rescue of anatomical and behavioral phenotypes related to the serotonin system in the BDNF
Met/Met mice.
An unexpected finding in our study was that fluoxetine administration at P21–P42 failed to increase hippocampal BDNF levels in BDNF
Met/Met mice (see Figure S2D in the
data supplement), whereas it robustly increases BDNF levels in wild-type and BDNF-HA reporter mice (see Figure S2A–D). A previous report in BDNF Val66Met heterozygous mice showed that while there is a significant reduction in BDNF protein levels in the hippocampus of BDNF
Val/Met mice compared with wild-type mice, there is no effect of genotype on mRNA levels (
26). As the variant BDNF
Met polypeptide is inadequately trafficked into secretory vesicles (
23,
67), anterograde delivery of BDNF to the hippocampus from other brain regions could account for diminished total levels.
Another possible explanation for the effects of early-life fluoxetine in BDNFMet/Met mice could be that fluoxetine pharmacokinetics differ with age. However, previously it has been shown that continuous administration of a fixed fluoxetine dosage provides clinically relevant steady-state plasma drug levels in juvenile mice that do not vary significantly with age.