Large international consortia have undertaken highly successful efforts to unravel the genetic underpinnings for several of the major psychiatric disorders. For schizophrenia, a genome-wide association study (GWAS) was performed using >36,000 patients and >110,000 controls, which allowed for the identification of 108 loci reaching genome-wide significance (
1). Using a polygenic risk score, these loci accounted for an estimated 3.4% of the variance in schizophrenia liability, which is generally considered insufficient for implementation as a routine clinical diagnostic measure (
9). The largest effort to date for identifying rare coding variants was performed using roughly 2,500 cases and an equal number of controls, yielding no variants reaching genome-wide significance (
4; but see also
10). Multiple genes have been reported to exhibit an increased burden of rare de novo mutations among patients with schizophrenia, which disproportionately involve genes coding for proteins that govern synaptic function (
11–
13). For bipolar disorder, the largest GWAS reported included a total of approximately 12,000 patients and 52,000 controls, in which genome-wide significant associations were observed for
CACNA1C and
ODZ4 (
14). For major depressive disorder, no genome-wide significant loci were identified in a 2013 GWAS mega-analysis comprising
∼9,200 cases and
∼9,500 controls in the discovery phase and
∼6,800 cases and
∼50,000 controls in the replication phase (
15). However, consistent with the widely held assumption that the genomic architecture of many polygenic disorders will yield to analyses involving increasingly large cohort sizes, a recent GWAS of major depression involving a discovery cohort of
∼75,000 cases and
∼231,000 controls, and a replication phase with
∼45,000 cases and
∼106,000 controls, identified 15 genome-wide significant loci (
16).
Genetic studies of autism spectrum disorder (ASD) have yielded notably strong associations with rare monogenic syndromes, including fragile X syndrome, tuberous sclerosis complex, and Angelman syndrome (
17,
18). Investigations of idiopathic autism spectrum disorder have converged on de novo mutations as a frequent genetic determinant (
19). The most recent large-scale study (
2), involving 2,500 pedigrees, found that de novo coding mutations and copy number variants (CNVs) together explain approximately 30% of simplex ASD cases. The CNV results confirmed multiple previously identified susceptibility loci for ASD, including
1q21.1,
3q29,
7q11.23,
16p11.2,
15q11.2–13, and
22q11.2 (
20). Notably, de novo point mutations and CNVs were also recently demonstrated as the major cause of severe intellectual disability (
21). Moreover, neurodevelopmental abnormalities, congenital heart disease, and extracardiac congenital anomalies appear to cosegregate among patients with de novo mutations (
22), together providing further evidence of pathogenic rare genetic variation in syndromic forms of psychiatric illness that involve comorbidity between axis I psychiatric disorders, intellectual disability, congenital abnormalities, and dysmorphic features.
Copy Number Variants
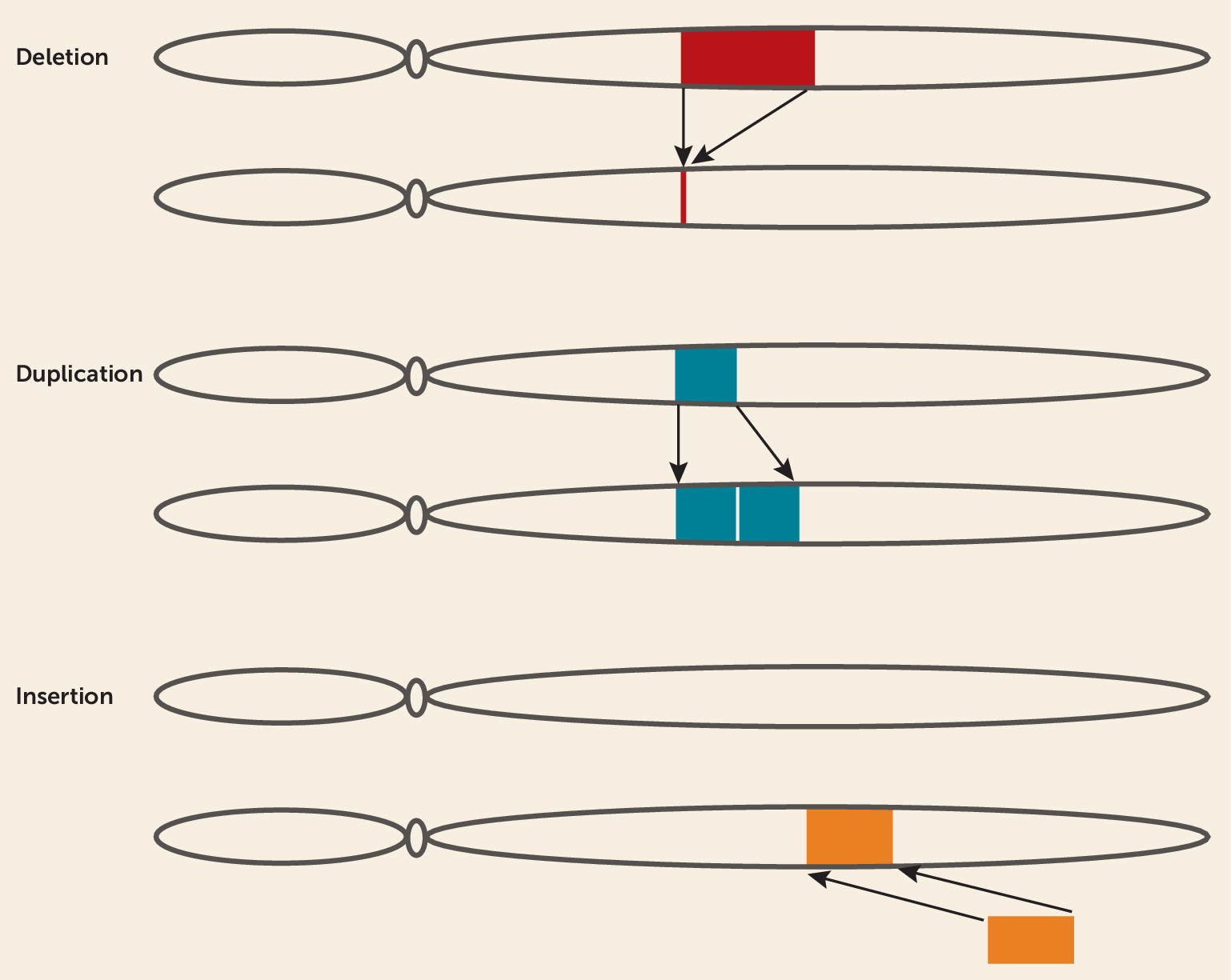
CNVs are a common source of structural genomic variation involving gains (duplications or insertions), losses (deletions), or complex rearrangements of genomic sequence resulting in deviations from the diploid state (
23,
24) (
Figure 1). On average, each individual is estimated to have >1,000 CNVs, with similar rates of gains and losses, together involving
∼10 Mb of genomic sequence (
25). CNVs can include one or more genes, leading to disruptions of coding regions or alterations of gene dosage, although many CNVs involve exclusively intergenic sequence. A wide variety of CNVs have been shown to be important sources of pathogenic mutations, particularly those of large size (>100 kb), low frequency (<1% in the general population), and containing genes (
5,
21,
25–
27). With the advent of microarray technology, reliable detection of CNVs has been widely implemented for both research and clinical diagnostic use in a standardized and relatively low-cost workflow (
25).
Rare CNVs have been well demonstrated as enriched in patients with a variety of severe psychiatric disorders (
5,
28–
30). The CNV and Schizophrenia Working Groups of the Psychiatric Genomics Consortium and the Psychosis Endophenotypes International Consortium recently published a collaborative study (
5) involving a large cohort of patients with schizophrenia (N=21,094) and unaffected controls (N=20,227). Overall, patients with schizophrenia were found to have a significantly increased burden of CNVs (>10 kb in size with a population frequency <1%). Eight loci reached genome-wide significance for their pathogenicity: deletions at
1q21.1,
2p16.3 (
NRXN1),
3q29,
15q13.3,
16p11.2 (distal), and
22q11.2, as well as duplications at
7q11.23 and
16p11.2 (proximal). Remarkably, CNV deletions at the
22q11.2 locus were observed in 64 of 21,094 cases, compared to one of 20,227 controls (p=5.7×10
−18; odds ratio=67.7, 95% CI=9.3–492.8). Carriers of these rare pathogenic CNVs have a significantly higher risk of developing psychosis and a range of other neuropsychiatric disorders, including mood and anxiety disorders, attention deficit hyperactivity disorder (ADHD) (
31), ASD (
32), and Parkinson’s disease (
33), as well as a variety of congenital malformations (
31,
34,
35).
The eight CNVs found to be significantly associated with schizophrenia are collectively present in 1.4% of patients and together explain 0.85% of the variance of schizophrenia liability (
5), while the 108 genome-wide significant loci of the latest schizophrenia GWAS explain 3.4% of the variance (
1). Although the relatively low prevalence of highly penetrant CNVs among patients with nonsyndromic schizophrenia currently appears to limit the cost-effectiveness of CNV testing in the standard psychiatric diagnostic workup, with increasingly large data sets and continued reductions in cost, this remains a distinct possibility in the future.
Compared to patients with nonsyndromic forms of psychiatric illness, those with syndromic features have a significantly higher frequency of CNVs. In one of the first studies to examine CNVs in patients with syndromic psychiatric illness, deletion of
CNTNAP2 was found to be associated with comorbid schizophrenia and epilepsy (
36). A subsequent study described patients with comorbid schizophrenia and epilepsy, in which 4% of the cases had a
15q11-q13 duplication while none were found in controls (
37). Similarly, in a study investigating the relative frequency of CNVs in schizophrenia patients with or without intellectual disability, an excess of large (>1Mb)
15q11.2 duplications or deletions was found in patients with schizophrenia and comorbid intellectual disability (
38). In an exemplary case, the
15q11.2 duplication carried by a proband was found to cosegregate in his family with schizophrenia and comorbid intellectual disability, hearing impairment, and ophthalmological problems.
The Clinical Genetic Examination and Consultation
Clinical geneticists provide a diagnostic service and counseling for individuals or families with, or at risk for, conditions that may have a discernible genetic etiology. Ideally, a clinical genetics consultation for patients with severe psychiatric disorders is an opportunity to assess etiology, prognosis, and risk for offspring through probability estimates based on empirical genetic findings (
8,
39,
40). In certain instances, clinical, molecular, or metabolic diagnostics can provide insight into the genetic cause of the disorder or syndrome (
19,
22,
41). Client-centered psychiatric genetic counseling for patients and their families has already been implemented successfully in a few specialized centers, with demonstrable enhancement of patient empowerment and self-efficacy (
39). Moreover, prenatal genetic counseling has also been demonstrated to be an important opportunity for discussing psychiatric illness risk, facilitating insight, and increasing etiological understanding in a therapeutic and collaborative manner (
40).
When a patient is referred for genetic consultation, the proband, parents, and/or spouse are typically interviewed to learn about the motivation for their visit. In addition to the anamnesis, a detailed family history is obtained with a particular focus on pregnancy, delivery, and early developmental milestones. The clinical geneticist performs a physical examination of the proband, including dysmorphology assessment, in order to evaluate for the presence of syndromic features and/or minor physical anomalies (MPAs) that might be suggestive of genetic disease. MPAs are congenital anatomical defects (e.g., deformities of the head, eyes, ears, mouth, palate, hands, and feet) thought to be indicative of abnormal ectodermal development during the first and/or second trimester (
42). Since the CNS is of ectodermal origin, MPAs are generally viewed as potential indicators of abnormal CNS development (
43). A large number of studies have documented an increased prevalence of MPAs in patients with psychiatric disorders of a presumed neurodevelopmental etiology, such as schizophrenia and ASD (
44).
In the present study, we specifically focused our investigation on patients with a syndromic form of psychiatric illness, defined as an axis I disorder in combination with multiple congenital abnormalities and/or dysmorphic features. A total of 50 patients with syndromic psychiatric illness, comprising two independent prospectively recruited consecutive case series, were screened for CNVs. In total, we identified pathogenic or likely pathogenic CNVs in 24.0% of patients (95% CI=12.2–35.8), with analogous findings in each independent cohort.
Method
Cohorts
Pilgrim Psychiatric Center cohort.
Medical ethical approval was obtained from the institutional review board of Pilgrim Psychiatric Center in Brentwood, N.Y., and the Mt. Sinai School of Medicine in New York City. Patients, none of whom had previously undergone any genetic testing, were evaluated between February 2000 and June 2001. Informed consent was obtained from all patients. Patients with schizophrenia or schizoaffective disorder were diagnosed according to DSM-IV-TR criteria using the Comprehensive Assessment of Symptoms and History (
45). Intelligence testing was performed using the Wechsler Adult Intelligence Scale, 3rd ed. (WAIS-III) with the relevant classification terminology proposed by Groth-Marnat (
46): average (full-scale IQ, 90–109), low average (full-scale IQ, 80–89), well below average (full-scale IQ, 70–79), and lower extreme (full-scale IQ, ≤69). Medical chart review, family history, dysmorphology assessment, and physical examination were performed by a qualified psychiatrist (J.I.F., S.M.).
Erasmus University Medical Center cohort.
Medical ethical approval was obtained by the institutional review board of the Erasmus University Medical Center in Rotterdam, the Netherlands. Patients were referred by their treating psychiatrist to the Department of Clinical Genetics for diagnostic evaluations between January 2012 and July 2013. All patients were diagnosed according to DSM-IV-TR using the Structured Clinical Interview for DSM-IV Axis I Disorders. Medical chart review, family history, dysmorphology assessment, and physical examination were performed by a qualified medical geneticist (A.J.A.K.). The parents of the patients were invited to provide DNA samples, but participation was declined in all but one case.
Presentation and History
For the purposes of this study, we operationally defined a “syndromic” presentation as a DSM-IV-TR axis I psychiatric disorder in combination with at least two dysmorphic features and/or congenital abnormalities involving the head, hair, face, chin, eyes, ears, nose, mouth, lips, teeth, neck, thorax, abdomen, genitalia, skin, extremities, stature, or spine.
Family history of psychiatric illness was obtained by constructing a three-generation pedigree for each proband and systematically interviewing the proband and available family members regarding the psychiatric history of each person represented on the pedigree.
Genotyping
DNA was extracted from venous whole blood. Genotyping of the Pilgrim Psychiatric Center cohort was performed using the Affymetrix GeneChip Human Mapping 250K Nsp Assay, with the Affymetrix GeneChip Command Console (Affymetrix, Santa Clara, Calif.) for quality control analysis and variant calls. Genotyping of the Erasmus University Medical Center cohort was performed using the Illumina HumanCytoSNP-12v2.1 microarray, with the Illumina iScan Control and GenomeStudio software program, version 2.1, as well as the Nexus Copy Number Discovery software program, version 5.0 (BioDiscovery, Hawthorne, Calif.). Deletions were considered if they were supported by more than five sequential probes and larger than 150 kb. Duplications were considered if they were supported by more than seven sequential probes and larger than 200 kb. The results were filtered to remove common CNVs (population frequency >1%) present in the UCSC Genome Browser (hg18 build), the Database of Genomic Variants, and in-house databases. Copy-neutral regions of homozygosity were considered if they were larger than 5 Mb. Fragile X Syndrome testing was performed in the Erasmus University Medical Center cohort by Southern blot analysis of the FMR1 trinucleotide repeat length.
Pathogenicity Classification
CNV pathogenicity classification was implemented according to the guidelines of the American College of Medical Genetics (
47) through consensus between a molecular geneticist and clinical psychologist (C.G.B.) and a medical geneticist (A.J.A.K). In rare instances of discordance regarding variant classification, a consensus decision was made through a collaborative discussion together with a qualified psychiatrist (S.A.K.). Variants were classified as pathogenic if there were at least two published articles in the literature describing the variant as being associated with the proband’s phenotype. All other CNVs were classified as variants of uncertain significance (VUSs), with the specifier “likely pathogenic” assigned when there were at least two published fundamental neurobiological, functional genomic, and/or human genetic studies involving genes contained within a given CNV for which there was evidence for a causal influence on disease-relevant neurodevelopment, neural circuit function, or behavior.
Metabolic Studies
Erasmus University Medical Center patients were screened for metabolic abnormalities in blood plasma and urine. Blood plasma fraction was isolated according to standardized clinical protocols from venous whole blood collected using lithium heparin–coated blood tubes. Metabolic diagnostics included the following: acylcarnitines, amino acids, bile acids, creatine, guanidinoacetate, homocysteine, homogentisic acid, imidazole compounds, methylmalonic acid, mucopolysaccharides, oligosaccharides, organic acids, orotic acids, phenylalanine, tyrosine, phytanic and pristanic acid, purines and pyrimidines, sialic acid, sialotransferrins, sugars and sugar alcohols, sulfatides, tetraglucoside, and very long chain fatty acids.
Discussion
We performed comprehensive genetic analyses in two cohorts of patients with a syndromic presentation of psychiatric illness. In 12 patients (24%), we identified a pathogenic or likely pathogenic genetic variant. We found a similar percentage of pathogenic or likely pathogenic CNVs in each independent cohort (Pilgrim Psychiatric Center cohort: 5/19, or 26.3%; Erasmus University Medical Center cohort: 7/31, or 22.6%). Among the identified CNVs, many have been previously established as known genetic risk factors for psychiatric disorders (22q11.2 microdeletion, 16p11.2 microdeletion, XYY syndrome) and developmental delay (3q13.31 microdeletion, 1q21.1 microdeletion). Furthermore, we observed likely pathogenic CNVs involving only a single gene that have been strongly linked to psychiatric illness (CNTN4, CNTN6).
Pathogenic CNVs associated with psychiatric phenotypes have a broad range of penetrance, varying from
∼2%−33% for bipolar disorder, schizophrenia, or ASD, while the exposed attributable risk for pathogenic de novo CNVs varies between 79% and 87% (
63). In particular, among individuals carrying a microdeletion at
22q11.2, the lifetime prevalence of psychotic disorders is
∼30% (
64). Increasingly precise penetrance estimates will greatly facilitate discussions between patients, their families, and their health care providers regarding diagnosis, treatment, and prognosis (
25).
Our findings are consistent with a recent study by Stobbe et al. (
65), who reported clinical diagnostic findings in 24 consecutively evaluated adult patients with syndromic ASD, of whom 20.8% were found to have pathogenic or likely pathogenic CNVs. Furthermore, our observed rate of pathogenic CNVs (7/50, or 14.0%) is significantly higher than in the general-population schizophrenia cohort reported by Rees et al. (
28) (171/6882, or 2.48%) (Fisher’s exact test, p=2.6×10
−4). Therefore, clinical genetic testing for CNVs may be particularly relevant in patients with syndromic forms of psychiatric illness.
Clinical genetic testing should be considered in the presence or absence of a significant family history. Although a positive family history suggests the presence of an inherited genetic variant, a negative family history can be indicative of a de novo variant. Notably, however, the absence of an identifiable inherited or de novo genetic variant does not rule out more complex genetic events due to somatic mosaicism that might not be detectable in DNA isolated from peripheral blood (
66,
67). The considerable diagnostic importance of de novo mutations has been firmly established for intellectual disability (
19,
21,
68). Accordingly, this might also be an important genetic mechanism underlying syndromic forms of psychiatric illness that should be evaluated in future studies. Moreover, future investigations with larger cohorts should be conducted to determine the relative contribution of the severity of intellectual disability, distinct congenital abnormalities, craniofacial dysmorphologies, and seizure disorders to the prior probability of CNVs in patients with psychiatric disorders.
Important clinical benefits have been shown to result from a genetic diagnosis. Multiple studies have reported a preference on the part of patients and their families to obtain an etiological genetic diagnosis, the benefits of which include improved knowledge of their disease and a feeling of empowerment to better advocate for themselves (
39,
69). Patients who receive a genetic diagnosis experience a strengthening of the therapeutic alliance with their psychiatrist and mental health care providers (
70). In addition, having an etiological genetic diagnosis facilitates access to medical benefits, educational opportunities, and social services for patients and their families (
71,
72). Accordingly, formal genetic counseling is a critically important opportunity for patients and their families to understand the risks and opportunities for reproductive planning (
39,
40).
Confirmation of a known genetic syndrome can also improve diagnosis, prognosis, treatment, and prevention. The knowledge of a genetic diagnosis provides a unique opportunity to consider future clinical course and prognosis on the basis of published information regarding patients with comparable genetic diagnoses. For example, CNVs involving
CNTNAP2 have been reported to increase the risk for adult-onset mutism (
73) and epilepsy (
36). Furthermore, pathogenic CNVs associated with neuropsychiatric symptoms frequently involve medical comorbidities for which diagnostic screening and preventive treatment are often available and of significant clinical consequence. For example, patients with
22q11.2 microdeletion exhibit high rates of immune deficiency, congenital heart disease, hypocalcemia, seizures (often provoked by episodes of hypocalcemia), scoliosis, obesity, hearing loss (sensorineural and/or conductive), thrombocytopenia, thyroid dysfunction, and renal anomalies (
74). Additional examples include
16p11.2 microdeletion, which is associated with obesity, gastrointestinal symptoms (e.g., reflux, constipation, diarrhea), seizures, immune deficiency, scoliosis, and congenital heart disease (
75,
76).
1q21.1 microduplication is associated with cardiac abnormalities (in particular tetralogy of Fallot), seizures, and macrocephaly (
77).
Regarding treatment, there are important examples of CNV-associated risks of adverse effects or complications of psychopharmacological treatment, as well as emerging therapeutic opportunities. For example, patients with schizophrenia and
22q11.2 microdeletion appear to be at increased risk of adverse events from clozapine, including seizures, movement-related side effects (including myoclonus, tremor, unsteady gait, rigidity, and slurred speech), neutropenia, and myocarditis (
78,
79). Clinicians should also be aware of the potential to exacerbate the predilection toward hypocalcemia in patients with
22q11.2 microdeletion through medication (e.g., anticonvulsants) or alcohol consumption, and the corresponding importance of vitamin D and calcium supplementation. Moreover,
16p11.2 microdeletion results in a strong predisposition to obesity, for which there should be concern regarding medications associated with weight gain and metabolic syndrome (
75,
80). With the increasing success of psychiatric genetics, neuroscientists are gaining knowledge about the underlying neurobiological mechanisms of CNV-associated neurogenetic syndromes for which clinicians, patients, and their families should be aware of emerging clinical research studies. Notable recent examples of translational drug development studies include vigabatrin for
22q11.2 microdeletion syndrome (
81) and oxytocin for
CNTNAP2 deletion (
82).
Craniofacial abnormalities are known to be associated with alterations in brain development, consistent with the shared neurodevelopmental origin of the brain and the facial skeleton (
83). However, the issue is complex, given that there is a generally broad phenotypic spectrum associated with carriership of pathogenic CNVs (
84–
86). In an Icelandic population-based study of more than 100,000 people without a history of schizophrenia or autism,
∼1% were identified as carriers of pathogenic CNVs. Notably, although none of the people carrying these CNVs met traditional clinical diagnostic criteria for a neurodevelopmental or neuropsychiatric disorder, the majority exhibited a range of functionally significant cognitive and neuropsychological deficits (
87).
For both schizophrenia (
5) and autism (
20,
88), case-control studies have shown a clear excess of CNVs. In contrast, the genome-wide burden of CNVs does not appear to be increased for bipolar disorder or recurrent major depression (
89–
91) (however, there is strong evidence for an association of bipolar disorder and recurrent depression with duplications at
16p11.2 [
48,
89]). Notably, large (≥100 kb) CNVs—which we have focused on in the present study—generally appear to have larger effect sizes than small CNVs, but they are also significantly more rare. Accordingly, the contribution of smaller CNVs to phenotypic diversity in the general population remains less well studied, because of the requirement for increasingly large population-based cohort studies with detailed phenotypic data and higher-density genotyping (
25). Future studies using high-resolution variant detection by whole genome sequencing are expected to greatly facilitate the identification of smaller pathogenic CNVs and indels.
The majority of the patients in our cohorts had intellectual disability in addition to axis I psychiatric disorders, for which the nature of the causality of the genetic effects remains an open question. Specifically, it remains unclear to what extent a given CNV might exert its effects directly through endogenous neurobiological mechanisms and indirectly through impairments in cognitive reserve (
92) or social or emotional functioning. In this regard, it is notable that genome-wide CNV burden appears to be elevated in patients who have a psychiatric disorder and comorbid intellectual disability (
2,
93,
94). This finding might therefore suggest that CNVs can exert their influence on psychiatric disease risk indirectly through reduced intellectual capacity (
95). Conversely, the pattern of allelic pleiotropy observed for distinct CNVs and rare single-nucleotide variants suggests that the associated risk of psychiatric illness is likely to be independent of intellectual disability (
48,
96,
97).
With regard to the classification of CNV pathogenicity, it is important to be aware that there remains a considerable degree of subjectivity when implementing the consensus guidelines (
47). Classifications of pathogenic and benign are relatively higher confidence, given the stronger available evidence base required for these specifiers, while likely pathogenic variants and VUSs are dynamically evolving with the increasing availability of population-based genotyping data (e.g., the ExAC browser [
98]) and clinical genetic information (e.g., ClinVar [
99]), as well as the standardized implementation of genome-wide functional genomic, neuroimaging, and cognitive analyses (e.g., the ENIGMA consortium [
100]).
Despite the substantial benefits for patients and their clinicians, genetic testing is not without clinically significant risks. Given that genetic testing currently leads to an etiological diagnosis in only a minority of cases, patients’ expectations should be appropriately tempered. In the absence of identifiable pathogenic CNVs, there remains a distinct possibility that CNVs classified as VUSs and/or other types of genetic variants, such as rare protein-coding mutations or common polygenic risk, may be etiologically significant factors. Moreover, for patients who receive an inconclusive diagnosis (e.g., VUSs), the resulting uncertainty might increase their level of concern (
69). Conversely, a confirmed pathogenic finding may induce anxiety regarding future prognosis and later-onset symptoms (
70). Importantly, the decision of whether to perform genetic testing for diagnostic or predictive purposes should be made by the patient (or by a legal guardian, for those who are incapable of providing informed consent), and only after a comprehensive discussion with their psychiatrist and genetic counselor to weigh the risks and benefits of testing. Moreover, the outcome of genetic testing for a patient can have significant consequences for the entire family, for whom genetic counseling is strongly recommended to allow them a forum, either individually or as a group, regarding the implications for themselves and their children.
In summary, on the basis of the best available evidence, we propose that CNV screening should be considered for implementation within routine clinical practice for patients with syndromic forms of psychiatric illness. Although formal cost-effectiveness analyses are not yet complete, the current price of comparative genomic hybridization microarray testing is approximately $500–$1500 (
101,
102), with a number needed to test of 4.35 based on the combination of our results and those of Stobbe et al. (
65) (17 cases with pathogenic or likely pathogenic variants of a total of 74 screened). With the availability of exponentially larger data sets, an increasing proportion of the variants now considered likely pathogenic or VUSs on the basis of insufficient information are likely to be reclassified in the near future with even further improvements in the diagnostic yield for genetic testing of patients with syndromic forms of psychiatric illness.