Although our genomes have the remarkable capacity to transmit information across generations with high fidelity, there are a small number of regions flanked by seemingly innocuous repeat sequences that can misalign during replication and lead to deletions or duplication of large stretches of DNA (from several thousand to several million bases), commonly referred to as structural or copy number variation (CNV). A number of these susceptible regions, or “hot spots,” occur throughout the genome, leading to recurrent CNVs that are associated with syndromic presentations that have been recently dubbed “genomic disorders.” One of the first genomic disorders to be linked with neuropsychiatric symptoms is the 22q11.2 deletion syndrome (22q11.2DS), initially described by DiGeorge as a congenital syndrome of immunological (thymic aplasia), endocrine (hypothyroidism), and cranio-facial abnormalities (
1). However, subsequent studies have shown that the 22q11.2 deletion is associated with a broader and more diverse phenotype than was first apparent, often including mild intellectual disability, autistic features, prominent anxiety, and attention deficit symptoms. Most striking, however, was the recognition that approximately 25% of subjects with the 22q11.2 deletion manifest a psychotic syndrome indistinguishable from schizophrenia (
2). Indeed, in a recent study of more than 41,000 subjects (
3), the 22q11.2 deletion was found to be one of the strongest genetic risk factors for schizophrenia (odds ratio=67.7, 95% CI=9.3–492.8), being found in approximately 0.3% of cases with schizophrenia and almost no control subjects.
In this issue of the
Journal, a collaborative study by Bassett et al. provides important new insights that may help us understand why certain subjects with the 22q11.2 deletion develop psychosis while the majority do not (
4). This study reports initial findings from the International 22q11.2DS Brain and Behavior Consortium, a global consortium of researchers sharing data and collaborating to study the etiology, natural history, and treatment of individuals with the 22q11.2 deletion (
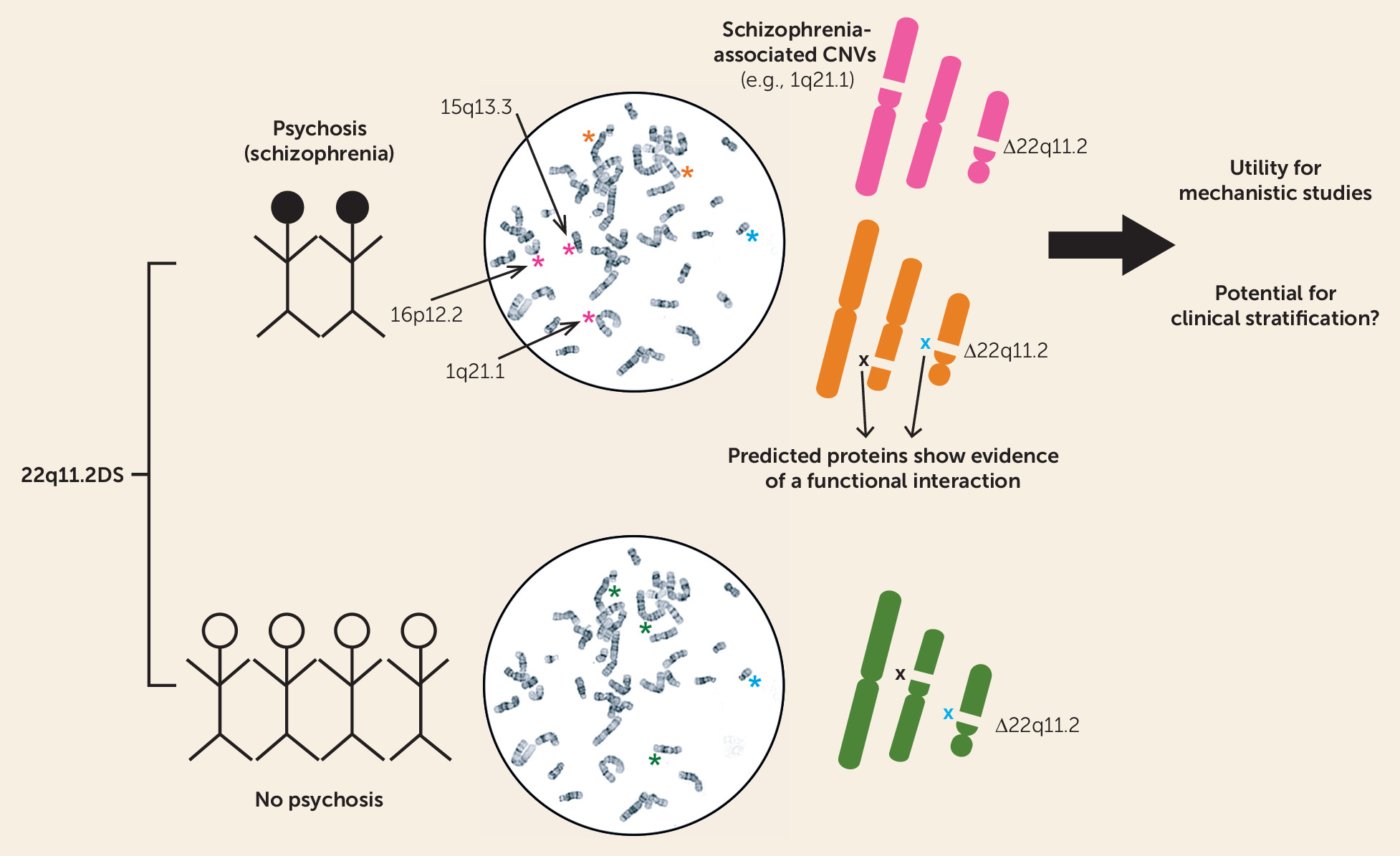
5). Bassett et al. hypothesized a role for “second hits” in the development of psychosis in subjects carrying the 22q11.2 deletion by investigating whether additional CNVs outside of the 22q11.2 region may increase the risk for developing psychosis or schizophrenia. Although there was no difference in the overall burden of CNVs between 22q11.2 deletions in subjects with schizophrenia and those without a history of psychotic symptoms, chromosomal microdeletions were significantly more likely to be protein-coding genes in the schizophrenia group compared with the group without psychosis (
Figure 1). Additional rare deletions in the schizophrenia group included 1q21.1 and 16p12.2 deletions, both of which have been associated with schizophrenia in general population samples. Cases with schizophrenia also had an excess of exonic duplications, particularly in genes associated with a nervous system phenotype gene set. Moreover, the proteins encoded by these enriched genes were more likely to be associated with proteins from the 22q11.2 deletion region, suggesting that the newly identified risk factors may act, at least in part, through a downstream pathophysiological mechanism similar to that of the primary 22q11.2 deletion.
However, as with any study, replication in sufficiently powered samples will be key to confirm the findings and may focus what is “enriched” among the cases with schizophrenia into a smaller number of genes or pathways that could be more feasibly tested in functional studies. As noted by the authors, despite the unprecedented effort to ascertain subjects with 22q11.2 deletions across the globe, the sample size is modest for genetic studies, particularly those looking at rare variants. Indeed, because many of the hypotheses tested by the investigators showed some enrichment without reaching statistical significance, future studies will need to confirm whether they may represent true positive associations in larger samples. A larger sample would also allow for a more comprehensive survey of additional genetic risk factors that may affect the penetrance or specific expression of neuropsychiatric phenotypes in patients with 22q11.2DS. Finally, although the present study focused on CNVs, it would be of interest to explore the implications of common and rare single nucleotide DNA variants in various clinical manifestations in patients with 22q11.2DS.
In the near term, replicated results would provide an essential entry point into biological and mechanistic studies with cellular and animal models that will be deployed in the study of the functional effects of the 22q11.2 deletion (
6). Because of its high penetrance and strong association in humans, the 22q11.2 deletion is likely to be one of the most promising leads for the study of disrupted cellular functions and circuits potentially associated with the emergence of psychosis in experimental models. Nevertheless, there are two major challenges. First, as the 22q11.2 deletion is associated not only with psychosis but also with several other psychiatric manifestations, we cannot simply assume that 22q11.2 deletion in cellular and animal models of the deletion are models for psychosis. However, by referring to other genetic disturbances that are estimated in silico to functionally interact with the 22q11.2 deletion for psychosis, we may be able to generate a more bona fide model for psychosis. Second, within the 22q11.2 deletion region, there are 46 protein-coding regions and 17 noncoding transcripts, any of which may potentially influence pathophysiology of the disorder (
7). To address this question, the genetic and functional networks identified by the present study may help shed light on which genes within the 22q11.2 region may be the more promising targets for biological studies.
Does this study have clinical implications? As described in the Treatment in Psychiatry article by Bouwkamp et al. in this issue of the
Journal, up to 24% of subjects with syndromic forms of psychiatric illness may harbor pathogenic or likely pathogenic CNVs (
8). For these subjects, a genomic diagnosis can have important consequences for treatment alliance, resource allocation, and appropriate management of medical and psychiatric comorbidities. Bassett and colleagues examined whether the enriched CNVs were associated with cognitive deficits but found no difference. However, different phenotypes (
9), such as morbid obesity, seizures, and cardiovascular complications, may merit further studies along the same lines. Moreover, as additional etiological risk factors are found, they may ultimately have the predictive ability to help identify the 22q11.2DS subjects who may be at highest risk of developing schizophrenia and who thereby are the most appropriate population for early intervention.