Psychotic symptoms occur in ∼40%–60% of individuals with Alzheimer’s disease (
1). The occurrence of psychosis in Alzheimer’s disease is heritable (
2), and it identifies a phenotype in which patients experience more rapid cognitive decline and elevated mortality compared with patients with Alzheimer’s disease without psychosis (
3). In addition, patients with psychotic symptoms in Alzheimer’s disease exhibit greater functional impairment, are more likely to be institutionalized during illness, and experience higher rates of additional neuropsychiatric disturbances, including aggression, agitation, and depression (
3). Current empirically developed treatments for psychosis in Alzheimer’s disease have limited efficacy, do not alter the more rapid disease progression, and are associated with substantial toxicity, including excess mortality (
3). Because the annual incidence of psychosis in Alzheimer’s disease is only ∼10% (
4), there is a window of opportunity to intervene to prevent psychosis onset. However, capitalizing on this opportunity first requires identifying the underlying neurobiology of resilience to psychosis in Alzheimer’s disease.
The hallmark pathologies of Alzheimer’s disease are fibrillar deposits of amyloid beta and phosphorylated tau. Studies have examined the association of these measures with psychosis in Alzheimer’s disease, and most have found that a lower phosphorylated tau burden is protective (
3). However, as a group these studies have several important limitations. For example, past investigations have overrelied on subjects with end-stage disease, despite clinical evidence that the most rapid increase in rates of psychosis in Alzheimer’s disease occurs in early to middle stages (
5,
6). In addition, studies often did not utilize uniform histologic procedures or unbiased quantitative measures of neuropathologic burden (
7). Importantly, additional core processes contributing to Alzheimer’s disease, such as neuroinflammation, microglial activation (
8,
9), and synapse dysfunction (
10,
11), have been largely unexamined in relationship to Alzheimer’s disease with psychosis. This omission is particularly glaring given that multiple neuroimaging studies have indicated relative preservation of neocortical synaptic structure and function in Alzheimer’s disease without psychosis relative to Alzheimer’s disease with psychosis, including greater perfusion (
12–
14) and metabolism (
15,
16) and higher gray matter density (
17). Finally, only a subset of studies have considered the role of common comorbid pathologies, such as the presence of Lewy bodies, 43-kDa TAR DNA-binding protein (TDP-43) inclusions, and vascular lesions.
Discussion
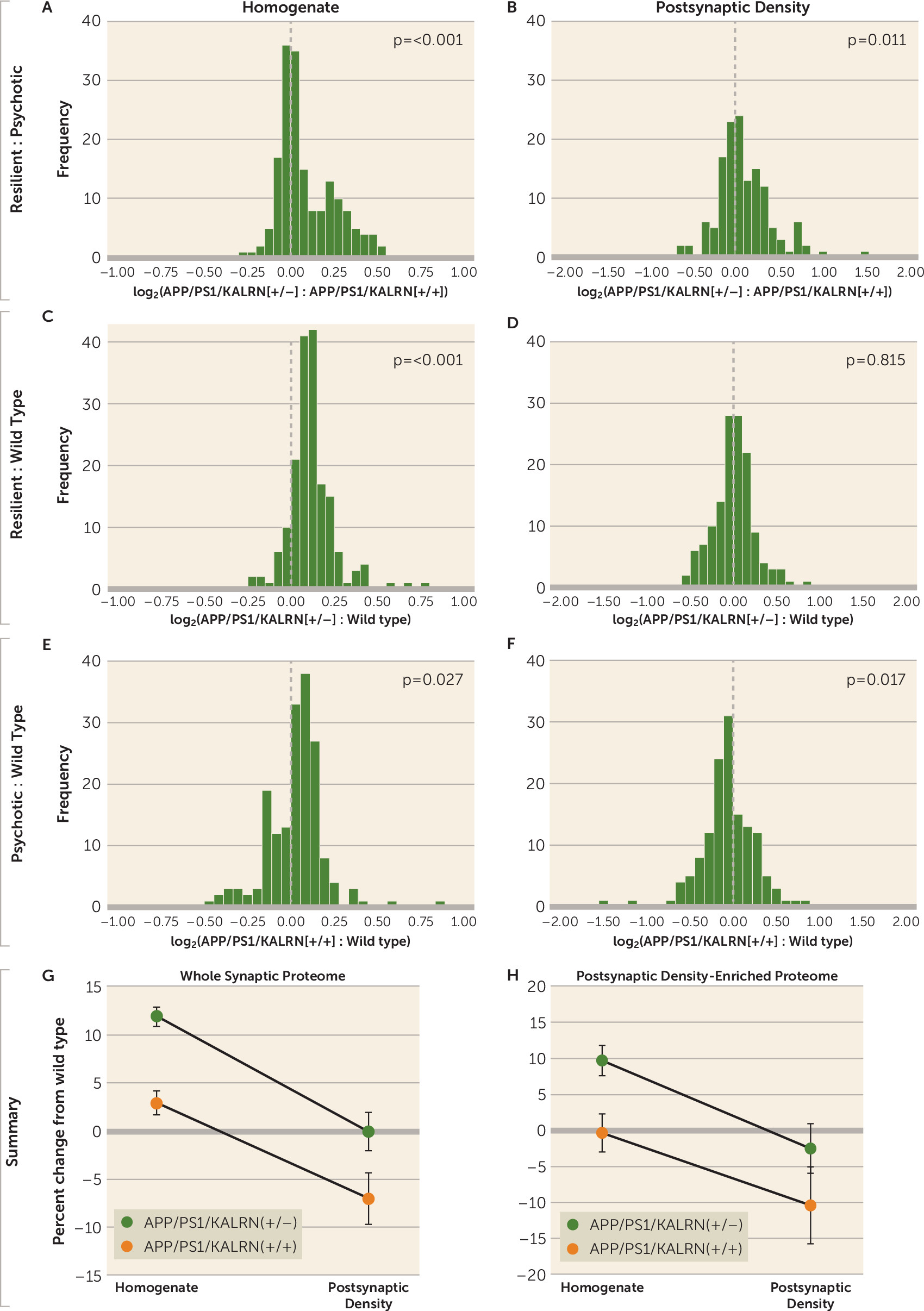
In this study, we combined quantitative neuropathology and LC-MS/MS to identify significant predictors of the resilient Alzheimer’s disease without psychosis phenotype. We detected significant contributions from PHF-1 tau burden and from the presence of TDP-43 pathology to the prediction of psychosis status in Alzheimer’s disease. Independent of these neuropathologic contributions, we then demonstrated that subjects with Alzheimer’s disease without psychosis have increased levels of synaptic proteins, particularly those that are enriched in the postsynaptic density, relative to those with psychosis. Finally, we recapitulated our human postmortem finding of elevated synaptic protein levels in Alzheimer’s disease without psychosis in a mouse model of amyloid beta overexpression that is protected from psychosis-associated behaviors that would otherwise progress between 6 and 12 months of age. Within the homogenate fraction, APP/PS1/KALRN(+/−) mice had elevated levels of postsynaptic density–enriched proteins compared with wild-type and APP/PS1/KALRN(+/+) mice, an effect that was associated with normalization of postsynaptic density–enriched proteins in the postsynaptic density compartment.
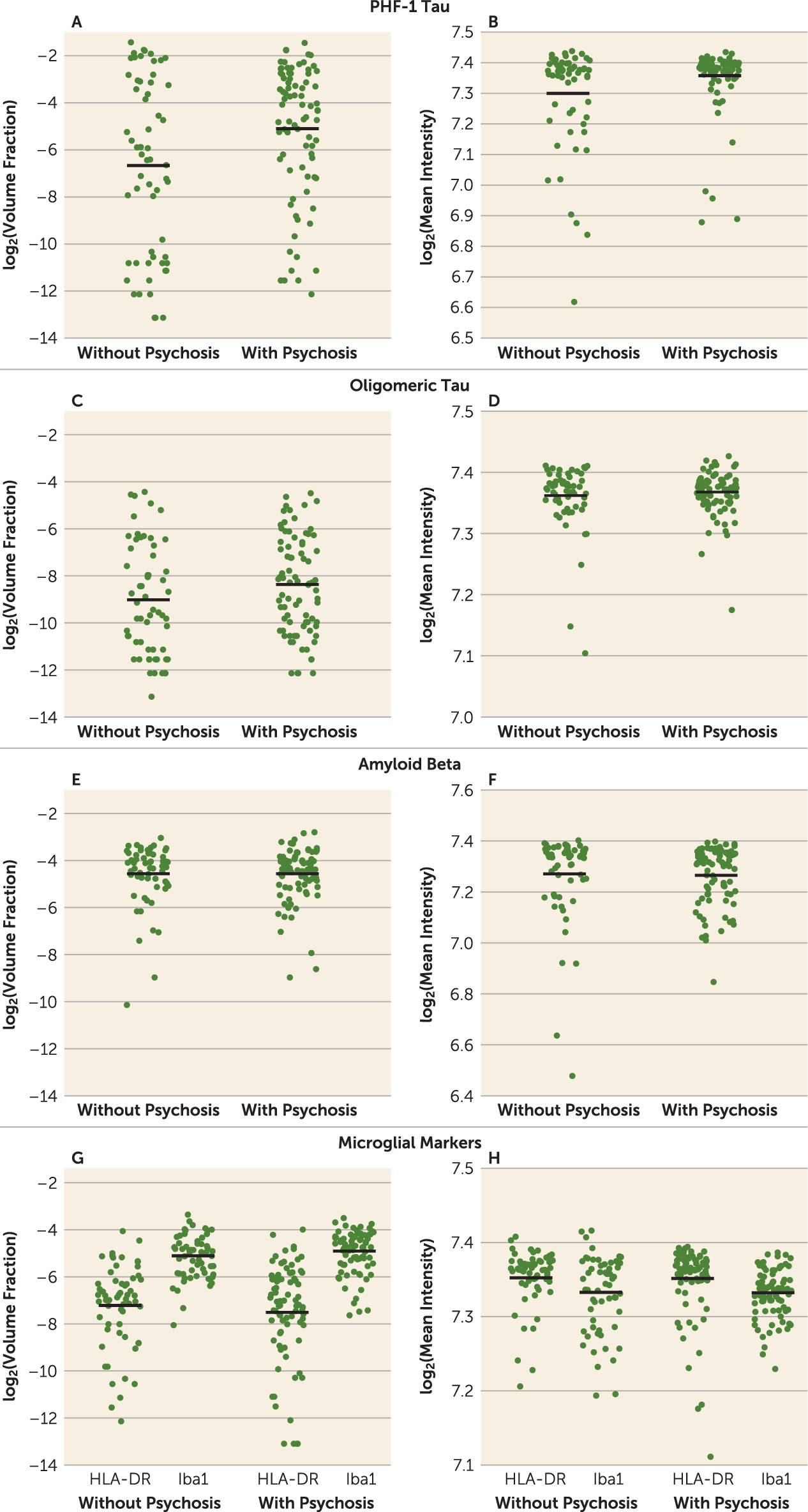
Previous studies exploring the neuropathologic correlates of psychosis in Alzheimer’s disease yielded variable results and were often limited by low case numbers, focus on single pathologies, or retrospective database review designs. To our knowledge, this study is the most comprehensive evaluation of neuropathologic factors to date, based on a large, well-characterized cohort from a single center and using unbiased quantitative approaches to measure disease burden. We confirmed the association of psychosis in Alzheimer’s disease with higher phosphorylated tau burden, as previously reported by our group and others (
3). While previous studies suggested that psychosis status was linked with cerebrovascular disease and microinfarcts (
23,
24), we did not detect a significant association between any of our vascular disease markers and psychosis in a logistic regression model. Contrary to previous reports, we detected less severe cerebrovascular disease in subjects with Alzheimer’s disease with psychosis in univariate analyses. The significance of this observation is unclear. We report, for the first time, a significant association between the presence of TDP-43 pathology and psychosis status. Concurrent TDP-43 is a frequent finding in brains of subjects with Alzheimer’s disease, seen in >50% of cases (
25). While the presence of TDP-43 has been associated with worse cognition and memory impairment (
25), no association with clinical subtypes has previously emerged (
26). Our study provides the first link of TDP-43 pathology with a specific clinical phenotype.
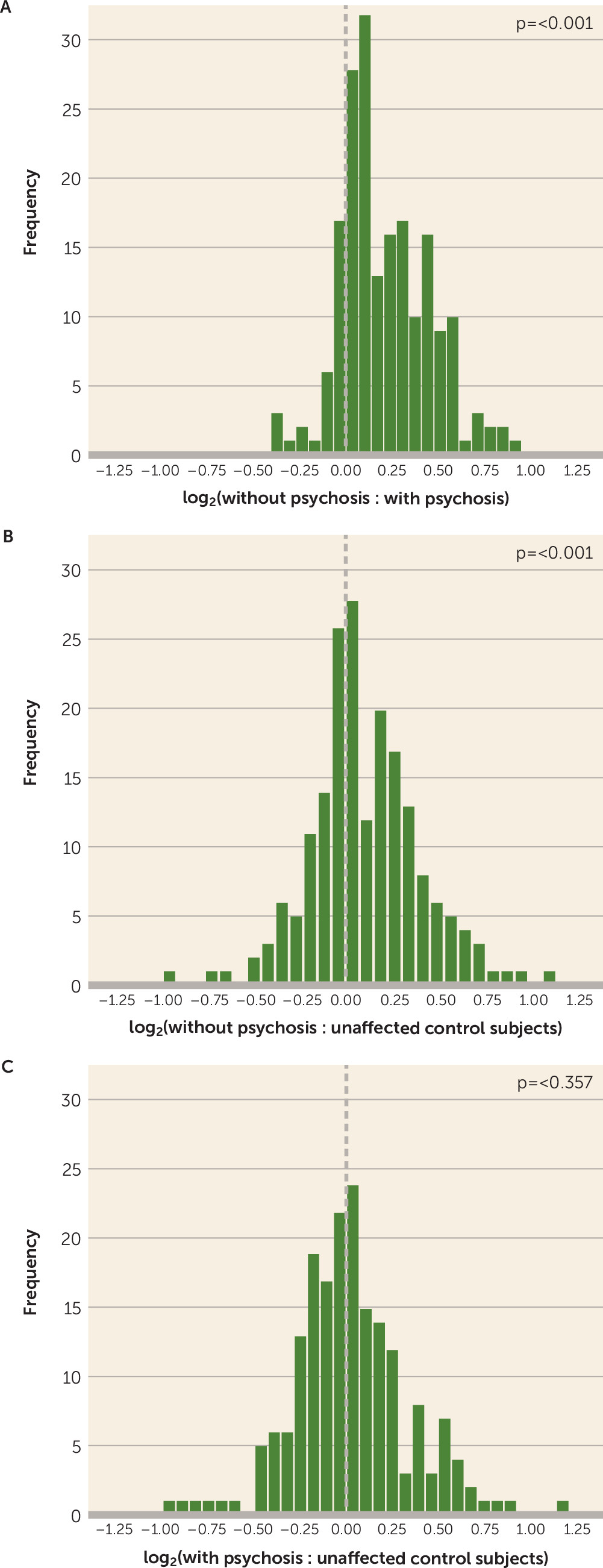
The presence of a compensated synaptic proteome has not, to our knowledge, been previously associated with psychosis status in Alzheimer’s disease. Perhaps the most replicated clinical correlate of Alzheimer’s disease without psychosis is a slower rate of cognitive impairment, and therefore our findings of synaptic proteome compensation in Alzheimer’s disease without psychosis are congruent with previous studies suggesting that clinically evident onset of Alzheimer’s disease is delayed by a period of synaptic compensation, during which cognitive ability is relatively maintained despite significant plaque and neurofibrillary tangle pathology (
27). Indeed, elevated synaptic markers during this period of mild cognitive impairment relative to both mild and severe Alzheimer’s disease have been described previously (
28). Others have demonstrated in hippocampal tissue that the synaptic proteome has a biphasic expression pattern across pathologic stage (
29), which is likely a compensatory effect early in disease. Further ultrastructural studies have suggested that there is morphologic evidence of compensation in the context of mild Alzheimer’s pathology, one of which demonstrated larger axospinous synapses within stratum radiatum and stratum lacunosum-moleculare, an effect that Nicholson and colleagues suggested represents increased synapse strength (
30). Our findings extend these earlier observations, revealing that synaptic compensations may persist into more advanced disease stages in a subgroup of individuals defined by the resilient Alzheimer’s disease without psychosis phenotype.
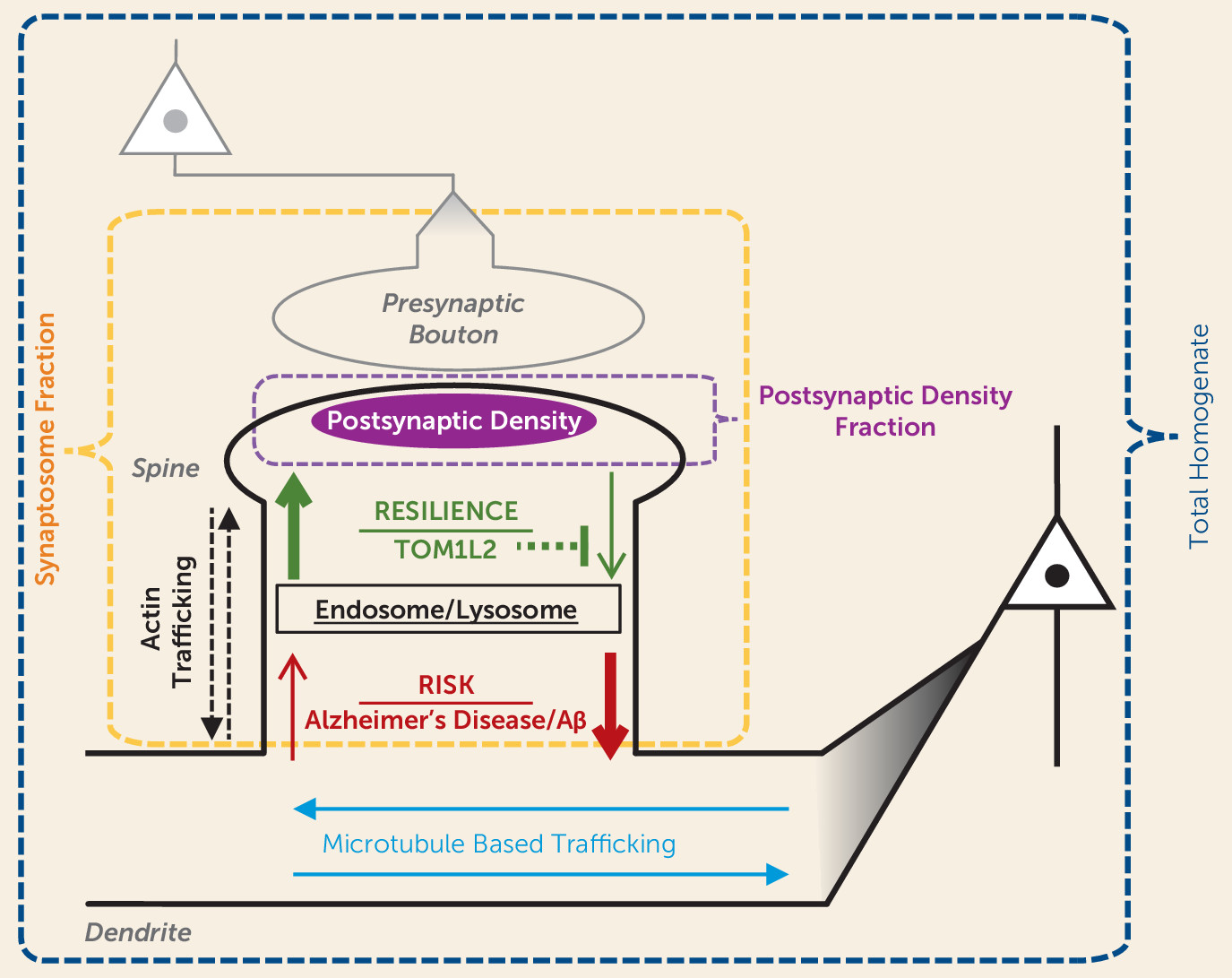
An important question is what mechanism(s) might underlie the synaptic proteome compensation in Alzheimer’s disease without psychosis. A model in which resilience to psychosis in Alzheimer’s disease results from a shift in the proteostasis of forward and back-trafficking of synaptic proteins to and from the postsynaptic density for function and degradation (
Figure 5) would be consistent with our data indicating that
Kalrn reduction protects the synaptic proteome in APPswe/PSEN1dE9 mice. Kalirin protein has been shown to regulate actin transport motors and trafficking of endosomes (
31). Indeed, we found evidence of accumulation of endocytic vesicle trafficking proteins in the homogenates of APP/PS1/KALRN(+/−) mice in conjunction with normalization of postsynaptic density protein levels. Furthermore, we recently identified a set of common single-nucleotide polymorphisms (SNPs) that are associated with resilience to psychosis in Alzheimer’s disease (
32). Among the consistently protective SNPs was rs8082590 on chromosome 17, the protective allele of which is associated with reduced expression of TOM1L2 (
33), the gene encoding Target Of Myb1 Like 2 Membrane Trafficking Protein. TOM1L2 protein is required for maturation of autophagosomes and subsequent fusion with lysosomes (
34). Autophagy pathways that regulate the turnover of endosomal material from the synapse have strong evidence for involvement in Alzheimer’s disease (
35). Therefore, reduced TOM1L2 expression conferred by a common SNP may be a novel mechanism for increased synaptic protein levels and resilience to psychosis in Alzheimer’s disease. Autophagy pathways have also been shown to modulate the phosphorylation state of tau (
36,
37), providing a putative link to the findings in this and previous studies of reduced fibrillar tau in Alzheimer’s disease without psychosis (
38) and others’ similar findings of reduced PHF-1 tau in synaptosomes from subjects protected from Alzheimer’s disease despite sufficient neuropathology for a postmortem Alzheimer’s disease diagnosis (
39). Studies that examine the synaptic, morphologic, and behavioral effects of reduced TOM1L2 expression may provide further insight into mechanisms underlying a resilient Alzheimer’s disease phenotype.
We have interpreted our synaptic findings as representing resilience in the context of equivalent pathologic stage, based on several methodologic considerations that deserve discussion. We used an automated immunostaining method to provide uniform measures of multiple pathologies across subjects. Nevertheless, mean intensity values for these pathologies were skewed to the right, an effect that was particularly evident for detection of PHF-1 tau intensity. This limitation may have hindered our ability to identify even greater increases in PHF-1 tau burden in our Alzheimer’s disease with psychosis cohort, possibly affecting the degree of correction of our synaptic measures for this pathology. In addition, we note that we did not use this parametric approach to quantify the presence of comorbid Lewy body and TDP-43 pathology, as the burden of these pathologies within dorsolateral prefrontal cortex was too low and would have resulted in nearly all subjects being quantified at floor values.
Another potential concern regarding our synaptic findings relates to the control cohort used for LC-MS/MS. First, the control group was significantly younger than our groups of subjects with Alzheimer’s disease with and without psychosis. This discrepancy resulted from our effort to obtain a neuropathologically unaffected cohort, minimizing the possibility of Alzheimer’s disease–related pathologies influencing the synaptic proteome within subjects who appeared clinically unaffected. Several considerations, however, make this unlikely to be cause for concern. A recent proteomics study of hippocampal tissue from subjects 22–96 years of age demonstrated an effect of age on only 60 of 4,582 proteins assayed; only two of these 60 proteins, hippocalcin and vimentin, were contained in our synaptic assay (
40). Moreover, we detected profound differences in global synaptic protein levels when only our precisely age-matched subjects with Alzheimer’s disease with and without psychosis were considered. A second concern is that postmortem intervals were longer in our control group. However, we previously demonstrated that the majority of the synaptic peptides assayed exhibit stable detection for periods longer than the postmortem intervals of our subjects (
22), and therefore the differences we observed are unlikely to be an effect of postmortem interval.
In summary, we have demonstrated that increased levels of synaptic proteins are associated with protection from a more severe Alzheimer’s disease phenotype, Alzheimer’s disease with psychosis. This putative compensation exhibited by the synaptic proteome was independent of burden from multiple neuropathologies. While we have identified and described an analogous mouse model of Alzheimer’s disease without psychosis, both behaviorally and synaptically, further studies of animal models of resilience are necessary to detect protective mechanisms for psychosis in Alzheimer’s disease. One such future model is advised by our recent genetic findings suggesting an association between reduced TOM1L2 expression and protection from psychosis in Alzheimer’s disease. Neuropathologic, synaptic, and behavioral assessment of this model may inform prospective development of therapeutic targets to confer resilience to psychosis in Alzheimer’s disease.