Autism is a lifelong condition that is characterized by greatly impaired social and communicative abilities as well as increased repetitive behaviors and stereotyped interests. It is a common heritable condition that affects as many as 1 in 42 males and 1 in 189 females (
1). Autism is an exceptionally heterogeneous behaviorally defined condition that may be parsed into severity-based subtypes (
2) but also may be best described as a continuum of symptom severity that progresses from minimal to substantial impact (
3). For example, as many as 31% of individuals with autism have an intellectual disability, but also as many as 44% of children with autism are identified as having average and above average IQ (
1). Similarly, traditional autism-like characteristics such as impairment in social reciprocity tend to follow a continuous distribution among individuals with autism (
4).
Autism can also be divided genetically into multiplex and simplex forms. The former requires that families have at least two affected siblings, and the latter requires only one affected child and one unaffected child. A growing body of data suggests that the two forms may represent distinct conditions and involve different genetic etiologies (
5,
6). Even though it has been suggested that simplex autism is more common (
7), the largest studies of autism reported so far do not appear to corroborate this view and point to autism as primarily an inherited condition (
8,
9).
Important genetic components of autism are strongly suggested by heritability estimates that range from 50% to 90% (
8,
10). However, despite the high heritability, few genetic markers of substantial effects have been identified that can explain a large portion of the condition. Genome-wide association studies have identified numerous single-nucleotide polymorphisms (SNPs) of very small effect (
11,
12), and whole genome-wide copy number variation studies have identified de novo regions of very large effect in simplex children, yet these events appear to be rare (
13,
14).
Despite extensive efforts to identify the genetic factors underlying autism, a substantial portion of the expected genetic contribution to the disorder has not yet been identified. This missing heritability and the heterogeneity of the condition have led to hypotheses of polygenetic effects involving hundreds to thousands of small-effect loci and to speculation that autism could be caused by numerous different alleles acting in summation or in a complex and unidentified pattern (
15). However, evidence suggests that this contribution may not be substantive (
16). Furthermore, few genetic studies have accounted for the extensive heterogeneity of the condition, and most label all affected as the same in classic case-control designs.
It has been proposed that the limited success of efforts to identify the genetic factors behind autism may be because the critical genomic contributors are being missed by currently available genomic technologies (
17). In this regard, highly duplicated, copy-number-polymorphic sequences are excellent candidates to fill such a role. Indeed, not only are such sequences missed by conventional genetic approaches, including exome sequencing, but also, because their copy number may vary continuously over a wide range, they have the potential to produce a broad spectrum of phenotypic variation.
One such candidate of high and variable copy number that has been absent from traditional genetic studies of autism is the Olduvai (formerly DUF1220) protein domain family (
18,
19), primarily encoded by
NBPF genes (
20). Olduvai domains are approximately 65 amino acids in length and show the greatest human lineage-specific increase in copy number of any protein coding region in the genome (∼300 copies in humans, with 165 being human specific) (
21,
22). Olduvai copy number also shows a striking correlation with brain size and neuron number among anthropoid primates, and with brain size and cognitive aptitude in the human population (
23–
25). Interestingly, most human Olduvai copies map to 1q21, a genomic region in which duplications have been linked with autism (and macrocephaly) and reciprocal deletions to schizophrenia (and microcephaly) (
26,
27).
Olduvai copies have been parsed into six primary evolutionary subtypes based on sequence similarity: conserved (CON1-3) and human lineage specific (HLS1-3) (
21). The primary means of adding human-specific copies has been via amplification of the Olduvai triplet: a three-domain unit composed of an HLS1, HLS2, and HLS3 subtype (
21). Olduvai copy number is also highly variable in the human population (ranging from 250 to 350 copies) but, because of the repetitive nature of the family, has not been directly studied in conventional genetic studies of autism. Hence, Olduvai sequences represent a rich source of unexamined functional allelic variation.
Using an earlier method to follow Olduvai copy number (droplet digital polymerase chain reaction, ddPCR), we previously demonstrated (
28) that in multiplex families the copy number of Olduvai subtype CON1 showed a linear association with the severity of the primary symptoms of autism, a finding that was subsequently replicated in a second population (
29). However, CON1 subtypes, which range from approximately 60 to 85 diploid copies in humans, are found at >20 dispersed loci on chromosome 1, and ddPCR only measures the total number of copies in the genome. As a result, these previous studies were unable to provide information on which copies may be critical to the associations with severity.
To address this limitation, we combined two recently developed resources: availability of whole genome sequences for large numbers of individuals with autism (
30,
31), and a higher-resolution method for measuring Olduvai copy number (
32). Using these tools, we report here a third linear association between CON1 dosage and the severity of the core symptoms of autism in an independent population. We also implicate gene-specific Olduvai subtypes as potential drivers of this association. Finally, we show that the Olduvai associations are preferentially found in multiplex as opposed to simplex families and thus may be important to inherited forms of autism.
Methods
In this study, we first sought to replicate previous work in an independent population using different assay methods, and once replication was identified, data from all three studies were combined for an overall evaluation. Next, a latent genotype was constructed from specific NBPF genes and then compared with a latent phenotype to identify important NBPF genes.
Replication Population
Individuals with phenotype data from the Autism Genetic Resource Exchange (
33) and whole genome sequence (WGS) data from the Autism Speaks MSSNG consortium (
30,
31) were used for the replication analysis. Individuals met autism criteria through the Autism Diagnostic Interview–Revised and the Autism Diagnostic Observation Schedule. While a large number of samples have been collected in the Autism Speaks MSSNG project, we were able to assemble 215 multiplex samples that were not included in our previous investigations and that had appropriate phenotype measures for replication. Nonwhite race/ethnicity was included and identified by analyses of WGS data using a consensus of Google-developed machine learning algorithms (see
https://cloud.google.com/blog/products/gcp/genomic-ancestry-inference-with-deep-learning) and the ADMIXTURE software tool on a subset of common SNPs (
34). Given the small numbers of specific minority groups, nonwhite and white ethnicity were collapsed into two categories. Population stratification was addressed through the addition of this race/ethnicity variable in multivariate regression analyses, as discussed further below.
Copy Number Measurement
Copy number of Olduvai domains in the replication population was obtained through a WGS read-depth method (
32). Briefly, the read depth of regions containing Olduvai domains was compared with the range of location of the regions based on hg38. The depth of reads compared with the breadth of the region of interest covered, corrected by GC content and copy number of ultraconserved sequences, yielded a total copy number measurement of candidate Olduvai sequences per individual. This technique was validated by ddPCR and is explained further elsewhere (
32). Details on all aspects of whole genome sequencing and mapping for all data are provided as part of the Autism Speaks MSSNG Project (
31). To ensure that cross-platform variation did not influence results, data from only one sequencing platform (Illumina HiSeq X) were included. Briefly, sequences were generated using paired-end reads of 150-bp length on an Illumina HiSeq X platform, following Illumina’s recommended protocol. Given high correlation among HLS1, HLS2, and HLS3 domains, we utilized HLS1 measurements as representative of HLS2 and HLS3. The original reports that identified severity associations relied on ddPCR-derived copy number that determined total genomic copy number, for example, for the CON1 subtype. Read-depth analysis of WGS data, on the other hand, allows specific subtype copy number to be identified within specific
NBPF genes (
32). In order to replicate previous work, we summed Olduvai CON1 copy number derived from WGS data across all
NBPF genes and used it as the primary explanatory variable in multivariate regression.
Replication Analysis
Linear multivariate regression with a random intercept for sibling family was used to test the association between total Olduvai CON1 and autism symptom severity. The mixed model was fitted with reduced maximum likelihood, and tests of significance were two-tailed t tests. Severity was determined by the social diagnostic score (SDS) and the communicative diagnostic score (CDS) from the Autism Diagnostic Interview–Revised (
28). Covariates included other Olduvai domain totals (CON2, CON3, and HLS1) as well as sex, age, Raven’s Progressive Matrices IQ, and race/ethnicity. Reduced models were developed through a backward selection procedure, keeping only significant covariates (p<0.05) and the suggestive interaction term sex by CON1 (SDS, p=0.10; CDS, p=0.02). The Akaike information criterion supported the use of a random effect with random intercept for sibling family. An interaction of CON1 by sex was explored, given the biological plausibility. Residual diagnostics did not indicate departures from linear model assumptions.
Combined Analyses
Data from the three projects were combined for final evaluation. Associations of CON1 and HLS1 copy number with symptoms were tested with multivariate linear regression, as previously described. Random effects, family ID, and study were explored but were not supported by the Akaike information criterion and therefore were not included. Backward selection with the appropriate previously mentioned covariates was utilized, and results from parsimonious models are presented. Interactions of copy number by sex and copy number by inheritance were examined, and interaction p values are presented where simplex effects were explored.
Latent Variable Analysis
We used partial least squares path modeling (PLS-PM) to explore contributions of specific
NBPF genes to a latent genotype and social severity phenotype. Bootstrapped mean estimates and 95% confidence intervals were generated with 5,000 iterations to validate PLS-PM associations. Gene and phenotype loading p values using z-statistics derived from the bootstrapped error were generated as recommended in other work (
35) and are presented to aid in the evaluation of multiple comparisons during the latent variable construction. A conservative critical alpha in this stage of analyses is a Bonferroni-corrected p value of 0.002 (0.05/30). This corresponded to 11 of the larger and variable
NBPF genes (
NBPF1,
4,
6,
8,
9,
10,
11,
12,
14,
19, and
20), by two clades (CON1 and HLS1) in addition to eight phenotype measures described below.
To avoid inflation of the association between latent variables, PLS-PM model reduction was based on high levels of multicollinearity among Olduvai copy number and 95% bootstrapped intervals of loadings that did not include 0. All analyses were conducted with R (
https://cran.r-project.org/), the nlme package (
36), and the plspm package (
37).
Phenotype Information
This project used the Autism Diagnostic Interview–Revised to provide the SDS and the CDS to replicate previous findings. The diagnostic scores contain numerous subquestions, each organized into four subdomains that measure relevant social and communicative behaviors. These subdomains are then summed through an algorithm to create the diagnostic scores (the SDS and CDS) and give clinical guidance in diagnosis. The four subdomains of the SDS include SOC1, SOC2, SOC3, and SOC4, with questions probing items such as responding to other children, emotional sharing, seeking comfort, and social smiling. The four subdomains of the verbal CDS included COM1, COM2V, COM3V, and COM4 and explore language items such as stereotyped utterances, pronoun reversal, and social use of language.
Results
Replication Population
A total of 215 multiplex individuals were available for the replication analyses.
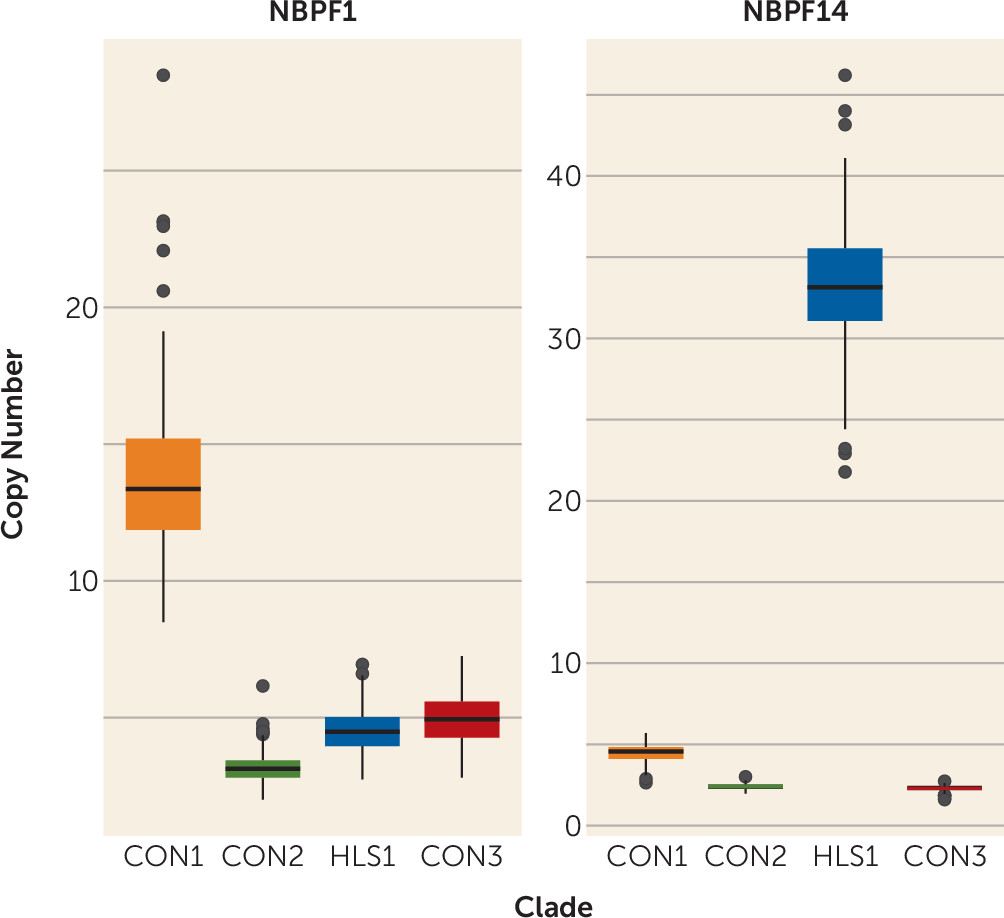
Table 1 provides a phenotypic description of the replication population, which was predominantly male (77%) and of Western European descent (77%). Sixty-eight sibling groups were used in these analyses, and group size varied from two affected siblings to four. The summed CON1 copy number ranged from 60.7 to 84.3 copies, with a mean of 70.7 copies, and followed a Gaussian distribution. Interestingly, CON1 copy number of
NBPF1 had substantial variation and ranged from 8.5 to 28.5 copies. HLS1 copy number of
NBPF14 was also variable and ranged from 22.9 to 46.2 copies (
Figure 1).
In the replication population, incrementally increasing copy number of total CON1 was associated with incrementally increasing SDS in males (β=0.25, 95% CI=0.02, 0.49; p=0.044) (interaction p=0.10) (
Table 2). A similar association was detected in males, where incrementally increasing copies of total CON1 were associated with incrementally increasing severity of CDS (β=0.23, 95% CI=0.21–0.25; p=0.008) (interaction p=0.02) (
Table 2). As presented in
Table 2, these results are highly similar to those in the previous two reports (
28,
29).
Combined Analyses
The combined population analysis included data for 524 individuals, with 64 simplex and the remainder multiplex. In multiplex individuals, for each additional copy of CON1, SDS severity increased 0.18 points (95% CI=0.08, 0.28; p=0.0008), and CDS severity increased 0.13 points (95% CI=0.05, 0.21; p=0.0009) (
Table 2) (SDS overall F test=5.16, df=4, 517, p=0.0004; CDS overall F test=6.11, df=2, 515, p=0.002). Age was a significant predictor (β=0.18, 95% CI=0.06, 0.30; p=0.002) in the SDS analysis. Increasing copy number of HLS1 was protective in the model of CDS (β=−0.02, 95% CI=−0.04, 0.0; p=0.046). The importance of multiplex over simplex was reaffirmed with SDS and CDS, where no associations were identified in simplex (SDS β=−0.05, 95% CI=−0.33, 0.21; p=0.72, interaction p=0.11; CDS β=−0.02, 95% CI=−0.23, 0.19; p=0.87, interaction p=0.16).
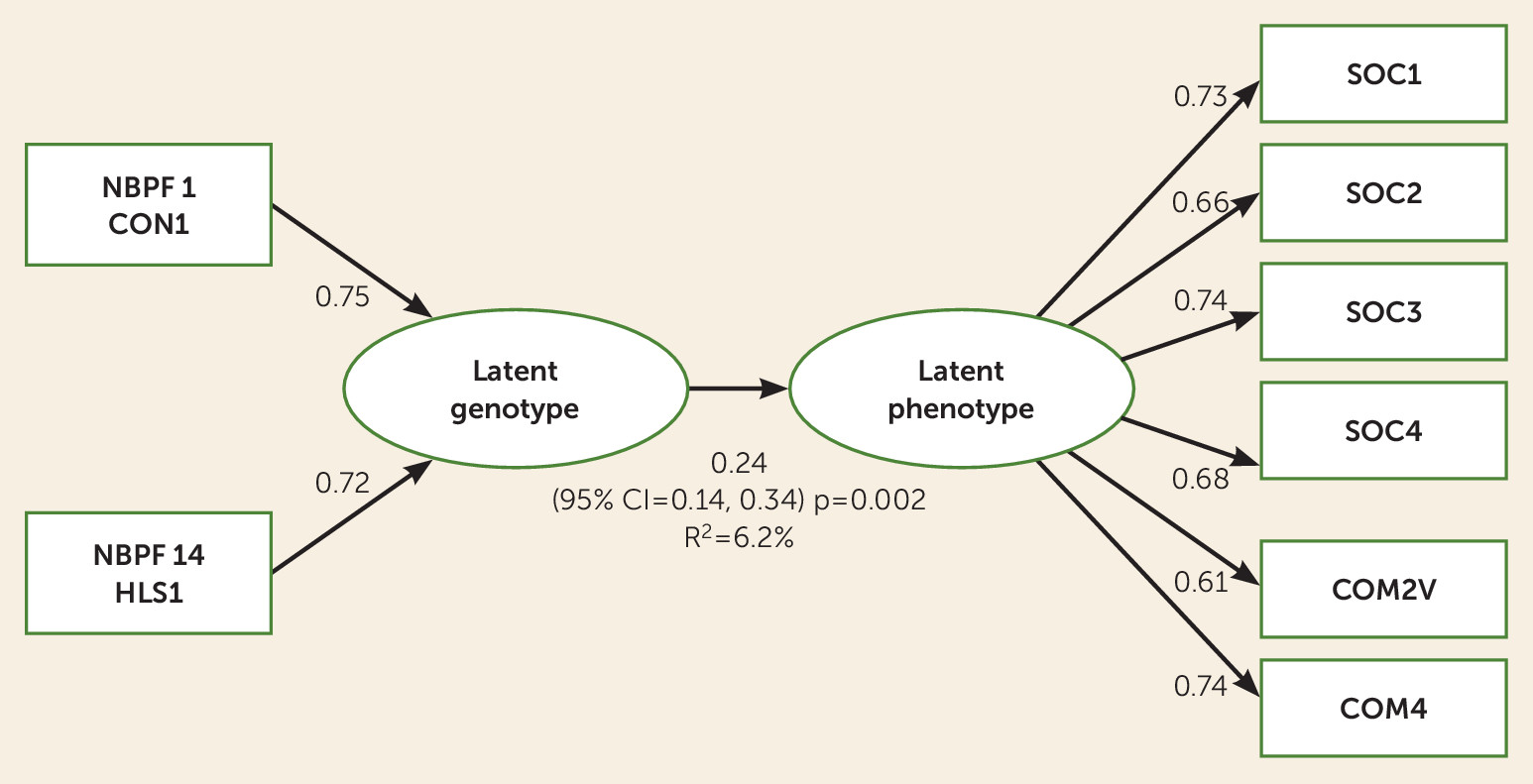
Latent Genotype and Phenotype Analyses
PLS-PM analysis identified an important latent genotype and latent severity phenotype that were associated with each other (β=0.24, 95% CI=0.14, 0.34; p=0.002), where the variation of the genetic latent variable explained a bootstrapped R
2 of 6.2% (95% CI=0.02, 0.12) of the variation of the latent social severity phenotype (
Figure 2). The communicative score domains and social domains had substantial positive loadings on the social latent variable, where SOC1, SOC2, SOC3, SOC4, COM2V, and COM4 were important (all p values <0.0001) (
Table 3).
NBPF1 CON1 (positive loading=0.75, 95% CI=0.19, 1.00; p=0.0004) and
NBPF14 HLS1 (positive loading=0.72, 95% CI=0.17, 1.00; p=0.0007) were important contributors to the latent genotype (
Table 3).
Discussion
Third Linear Association Between Olduvai Copy Number and Autism Severity
Using an independent population and a higher-resolution measurement method, we presented a third linear association between increasing Olduvai CON1 copy number and worsening of autism social and communicative symptoms. The patterns of associations we found here are highly similar to those previously reported (
28,
29), with only minor differences: the associations here are found predominantly in males and, compared with previous studies, show a larger effect size in communicative score. However, there are several reasons why such study-to-study variation can be expected: 1) autism is a complex condition with a wide range of phenotypic characteristics; 2) assessments of phenotypes can be expected to be imprecise, as they are primarily based only on caregiver reports (using algorithm scores from the Autism Diagnostic Interview–Revised) rather than physiological measurements; and 3) highly accurate copy number measurement of highly duplicated sequences, such as those encoding Olduvai domains, is difficult and copy number determinations are sometimes inexact.
We also showed that when the findings of this study were combined with those from our two previous studies, the linear association between CON1 and autism severity was unusually strong (N=524; β=0.18; SE=0.05; p=0.0008 for the SDS; and β=0.13; SE=0.04; p=0.0009 for the CDS). Given the substantial challenges related to accurate autism phenotyping and Olduvai genotyping, we believe it is remarkable that these studies have now identified similar associations in three independent groups. It is also noteworthy that, unlike in other recent genetic studies of autism, the associations reported here have been detectable, and replicable, using only modest sample sizes. Taken together, such findings strongly support a significant role for Olduvai CON1 in the severity of social and communication impairment in autism.
Associations Found Preferentially With Multiplex and Not Simplex Families
It is noteworthy that the significant linear associations with Olduvai copy number we report have appeared predominantly with multiplex as opposed to simplex families. This trend fits with other accumulating evidence that points to the possibility that multiplex and simplex autism may be genetically and mechanistically distinct disorders (
5). First, multiplex autism, which requires that at least two children in the family have autism, is thought to better represent the inherited form of autism. In contrast, simplex autism is often due to de novo events (
31,
38), but not exclusively so (
39). Second, the phenotypes of multiplex and simplex autism are typically distinct, with simplex tending to be more severe than multiplex phenotypes (
6,
40). Third, recent large-scale genome sequencing studies of simplex autism have not found genes that are involved in the typical core phenotypes of autism, such as social deficits and communication impairment. Rather, such simplex studies have preferentially found genes important to motor functions (
41). In contrast, our studies, now confirmed in three independent populations, suggest an opposite trend: we preferentially found associations involving the core autism phenotypes (social and communication impairment) in multiplex and not simplex families. In this regard, it is worth pointing out that early gene finding studies of inherited autism met with minimal success, a fact that has been mentioned as a reason for initiating genomic studies of autism using simplex families (
7). The findings we report here raise the possibility that the previous lack of success in identifying genetic contributors behind inherited autism may be because key sequences that contribute to multiplex autism involve difficult-to-measure, highly duplicated, dynamic, and copy number polymorphic sequences, such as those encoding the Olduvai family, and, as a result, have never been directly measured in other studies of autism (
17).
Candidate Olduvai Subtypes That May Drive the Associations With Autism Severity
In our previous studies, we reported linear associations between autism and the copy number of the CON1 subtype of Olduvai. However, the measurement methods we used could not determine which of the many copies of CON1 (approximately 60–85 diploid copies) were underlying the association. To address this limitation, we employed a high-resolution-sequence read-depth method that, when applied to WGS data sets, allowed many of the different NBPF genes and Olduvai subtypes to be discriminated from one another.
To identify specific NBPF genes and Olduvai subtypes that are important to a social severity phenotype, we examined our WGS read-depth data utilizing a latent variable analytic method, partial least squares path modeling (PLS-PM). Numerous two-by-two comparisons are commonly conducted in genetic association studies (or potentially in this instance, numerous one-continuous-gene-by-one-continuous-phenotype comparisons are possible). These comparisons assume independence, and they assume, moreover, that the genotype has a unique effect on a specific measured phenotype. However, in autism, many characteristics are highly correlated, making traditional two-by-two statistical approaches inefficient and redundant. At the same time, simply summing characteristics or clinically grouping other combinations of symptoms will reduce the number of comparisons but may not best describe the phenotype as it relates to an important genotype. PLS-PM addresses these challenges through the construction of the latent genotype and phenotype variables and then makes one statistical test between the two. In this case, of eight social metrics, two communication subscores were excluded and, of 22 NBPF genes and subtypes, two were retained—NBPF14 HLS1 and NBPF1 CON1. In this study, the latent genetic variable explained 6.2% of the latent social phenotype, which, in the context of genetic association studies of complex heterogeneous conditions, is considerable. This unusually large effect size speaks to both the importance of Olduvai in autism and the importance of refined phenotypes in complex conditions.
Potential Importance of NBPF14 HLS1 and NBPF1 CON1
NBPF14 HLS1.
The
NBPF14 gene is located in the 1q21 region of chromosome 1 and is part of a genomic sequence that has been repeatedly associated with autism (
26,
27).
NBPF14 is one of the four human
NBPF genes that together encode the great majority of human-specific Olduvai copies. These additional human-specific copies have been primarily generated by tandem intragenic expansion of the Olduvai triplet composed of an HLS1, HLS2, and HLS3 subtype (
21,
22), These subtypes show high sequence similarity, and because additions/deletions of entire triplets is the predominant form of copy number variation in the expanded
NBPF genes, the HLS1 subtype of
NBPF14 can be viewed as a proxy for a full Olduvai triplet. These three-domain blocks are also highly copy number polymorphic in the human population, among both typically developing individuals and individuals with autism (
28,
32). Thus, their high level of variation in humans, the inability of conventional genomic methods to directly examine these coding sequences (
17,
42), the multiple studies implicating this genomic region in autism (
43,
44), and our studies linking Olduvai copy number variation to the severity of the core symptoms of multiplex autism (
28,
29) make sequences encoding
NBPF14-related Olduvai triplets prime candidates for involvement in inherited forms of autism.
NBPF1 CON1.
The copy number of the
NBPF1 gene among humans appears to be highly variable, and the gene is thought to encode several CON1 domains (
21,
32). These characteristics fit with the idea that variation of CON1 in
NBPF1 has not been monitored in most genomic studies of autism, and that
NBPF1-related sequences (e.g.,
NBPF1L) may not be correctly mapped in the most recent human genome assembly (hg38). Given these observations, the possibility that
NBPF1 CON1 may be involved in influencing autism severity should not be considered surprising.
Genomic Trade-Offs and Links With Neurogenesis
The Olduvai protein domain family has been linked with both evolutionary benefit (copy increase is associated with brain expansion and cognitive aptitude [
23–
25]) and disease (increases have been linked to autism and macrocephaly and decreases to schizophrenia and microcephaly [
23,
45]). Such duality of effect has been incorporated into a cognitive genomic trade-off model that proposes that 1) Olduvai’s effect on brain size and neuron number may be dosage dependent, that is, more copies produce more neurons (
45), and 2) variation in Olduvai copy number can occur in different ways, and it is which, where, how, and when copies change that determines whether the consequences will be beneficial or harmful (
17).
Evidence suggests that the Olduvai trade-off may be related to variation in neurogenesis. For example, Olduvai has been shown to promote neural stem cell proliferation and exhibits a dosage-dependent association with gray matter volume and IQ in healthy individuals, both of which are related to neuron number (
23–
25). Olduvai copy number also shows a strong linear association with primate neuron number and brain size (but not body size) (
23,
24). Finally, human-specific Olduvai sequences have recently been linked with Notch-related pathways known to promote neurogenesis (unpublished data).
As previously suggested (
45), the involvement of Olduvai in influencing neuron number, possibly via centrosome-mediated effects, may also be relevant to its role in autism and schizophrenia. It is well established that how and when neurons are produced during brain development involve processes that, if altered, can produce atypical neuronal density and growth. In this regard, excesses in neuron number along with accelerated brain growth have been associated with autism (
46). In contrast, neuronal deficits may be important in schizophrenia, where smaller gray matter regions (
47) and reduced Olduvai copy number (
48) have been reported. In short, as Olduvai increases, autism severity and brain size increase, whereas the opposite trend is true for schizophrenia. These observations add further support to the proposal that autism and schizophrenia may be opposites of a cognitive disease continuum (
43) involving opposites in Olduvai dosage (
17). Such findings raise the testable hypothesis that the associations of Olduvai dosage with the severity of autism and schizophrenia are due to Olduvai-driven abnormalities in neurogenesis.
Finally, the data reported here, linking Olduvai sequences with autism severity, using a different means of assessing copy number and a different population, provide additional support for such a trade-off model. Because of technological limitations involving measurement of highly duplicated sequences, a more detailed description of the nature of Olduvai variation in humans, in either disease or nondisease populations, has not yet been reported. For similar reasons, it has been difficult to examine the potential involvement of Olduvai variation in autism risk. Given that much of the genetic contributors to inherited autism remain missing (
17), and given the possibility that multiplex autism may be due to a single dominant transmitted trait (
16), it is intriguing that variation in the ∼300-member human Olduvai family has yet to be fully examined in inherited autism. Continued improvements in genome sequencing technology that will allow such sequences to be studied with greater precision should permit these questions to be tested more rigorously in the future.
Acknowledgments
The authors gratefully acknowledge the resources provided by the Autism Speaks MSSNG project and the Autism Genetic Resource Exchange Consortium as well as the participating families.