Schizophrenia affects approximately 0.7% of the population (
1), is characterized by disturbances in cognition, emotion, perception, and thought, and has a severe impact on quality and length of life (
2). Around a third of patients experience treatment-resistant schizophrenia, a form of the disorder marked by severe functional impairment in which symptoms fail to respond adequately to at least two first-line antipsychotics (
3). Clozapine is the most effective treatment (
4) and the only medication approved for treatment-resistant schizophrenia. Despite extensive evidence supporting its effectiveness, clozapine remains underprescribed worldwide, including in countries with highly developed health services (
5,
6). A key factor limiting clozapine’s use is its potential to induce severe adverse drug reactions, including agranulocytosis, which occurs in up to 1% of patients and necessitates regular hematological monitoring (
7). Other adverse drug reactions, such as seizures, tachycardia, sedation, weight gain, and hypersalivation, have been associated with either clozapine dosage or plasma concentration (
8).
Clinicians routinely use clozapine levels to assess adherence and guide dosage in the management of both therapeutic response and side effects (
9). This is an important strategy, as adverse effects are the primary reason for clozapine discontinuation (
10). However, there is high interindividual variability in plasma clozapine concentrations at given dosages (
11), often as a result of the effects of concomitant medications (
12), which complicates titration and presents challenges for any research that aims to assess the relationship between dosage, efficacy, and adverse drug reactions. Guidelines for therapeutic drug monitoring indicate that clozapine plasma concentrations in the range 0.35–0.60 mg/L are optimal for response (
13), and concentrations higher than 0.60 mg/L have been linked to serious adverse drug reactions. Dose-response relationships between clozapine concentration and weight gain (
14) or sedation (
15) have been suggested, although this has not been seen for all adverse drug reactions (
8). The accurate prediction of clozapine plasma levels therefore has important clinical implications. Sophisticated models incorporating lifestyle habits and metabolic indicators can explain up to 48% of the variance in clozapine levels in large patient samples (
16,
17), but no individual factors other than age, smoking habits, and sex have been found to be of clinical value (
11).
The use of genetic approaches to identify biomarkers that explain individual variability in drug metabolism and response is the basis of the field of pharmacogenomics. Although this discipline experienced strong growth in the mid-2000s, fostered by successes related to cardiology and oncology (
18), translation of these results into clinical settings has proved challenging (
19). This has been particularly true in psychiatry (
20), and clozapine research in this area showcases some of the difficulties of carrying out robust pharmacogenomics studies. Clozapine is metabolized by the liver (
21) with first-pass metabolism, driven primarily by the CYP1A2 enzyme, producing norclozapine (
N-desmethylclozapine), a pharmacologically active compound that can reach up to 90% of the circulating concentration of clozapine (
22,
23). Other metabolites have been identified, such as clozapine-
N-oxide, formed by CYP3A4 (
22), and
N-glucuronides, which are secondary and tertiary metabolites produced by the UDP-glucuronosyltransferase (UGT) protein superfamily (
24). These enzymes are all implicated in other drug metabolic pathways (
25,
26) and have been the focus of the search for genetic variants associated with clozapine plasma concentrations. Previous studies have examined candidate polymorphisms in small (usually N<100) samples (
27,
28). Promising results have been reported for variants of the ABCB1 drug transporter (
28), but no finding has yet passed the threshold of genome-wide significance, which is now widely accepted as being required for robust association, even in candidate gene studies (
29).
Here we report the first genome-wide association study (GWAS) of plasma concentrations of clozapine and its metabolites. By applying modern statistical modeling techniques, we exploited information from over 10,000 metabolite concentration assays taken from a sample of nearly 3,000 patients with treatment-resistant schizophrenia. We identified genome-wide significant polymorphisms that delineate clozapine’s metabolic pathways, and we discuss their relevance to the clinical management of treatment-resistant schizophrenia.
Discussion
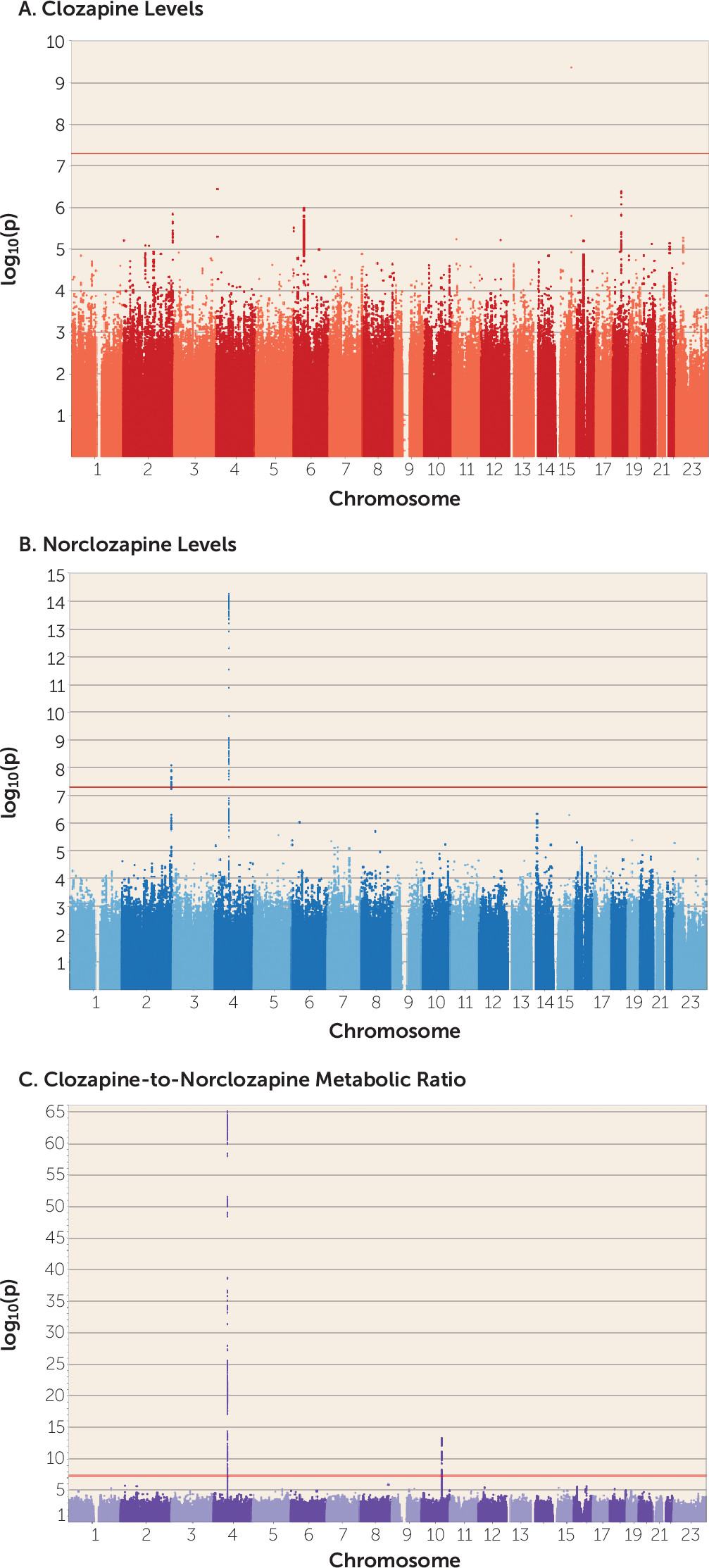
This is, to our knowledge, the first GWAS of clozapine metabolite plasma concentrations, which we carried out in 2,989 European individuals, the majority of whom had been assayed at multiple time points. Using statistical modeling to take advantage of all the available data, and a linear mixed-model GWAS approach, we provide the first robust evidence that alleles of specific
CYP and
UGT genes contribute to clozapine pharmacokinetics. This represents an advance from previous inconclusive studies (
28), mostly based on candidate marker surveys, and clarifies the relevance of common genetic variation in the proteins implicated in the clozapine metabolic route, which has been a matter of extensive debate. More specifically, our results support the hypothesis that the genetic architecture of clozapine metabolism may be driven by a few variants of large effect (see Figure S7 in the
online supplement), in line with other well-studied metabolic traits (
56).
The CYP1A1/CYP1A2 SNP rs2472297 associated with clozapine plasma concentrations lies in an intergenic region rich in binding sites for the aryl hydrocarbon receptor (AHR) protein, sites that are collectively known as “xenobiotic response elements” (
57). AHR binding is known to induce the expression of CYP enzymes in hepatocytes in response to the detection of many compounds, and thus variation in the regulatory function of AHR provides a strong candidate mechanism underpinning this association. While a causal association cannot be made solely on these grounds, disruption of normal AHR binding has also been suggested as explaining the association between variants at this locus and caffeine plasma levels, which may also influence coffee consumption (
51). Previous studies of clozapine levels and candidate polymorphisms at this locus have focused on common alleles within
CYP1A2 (
27,
28), none of which has been shown to influence its expression (
58). We also note that we find no support for other candidate genes from the literature, including the
ABCB1 variant rs1045642 (p=0.84), which was previously reported as associated with clozapine plasma concentrations in smaller (N<100) samples (
28). Both of these examples demonstrate the limitations of candidate SNP approaches that have been common in psychiatric pharmacogenomics to date and support genome-wide analysis to capture both coding and noncoding functional elements.
The results of our regression modeling show that the genetic modifiers of clozapine levels are comparable in impact to other known clinical and demographic variables, with their effect sizes being of the same magnitude as sex (
Table 1,
Table 2). An example to illustrate these effects and place them in clinical context is the observation that carrying one minor allele of rs2472297 at
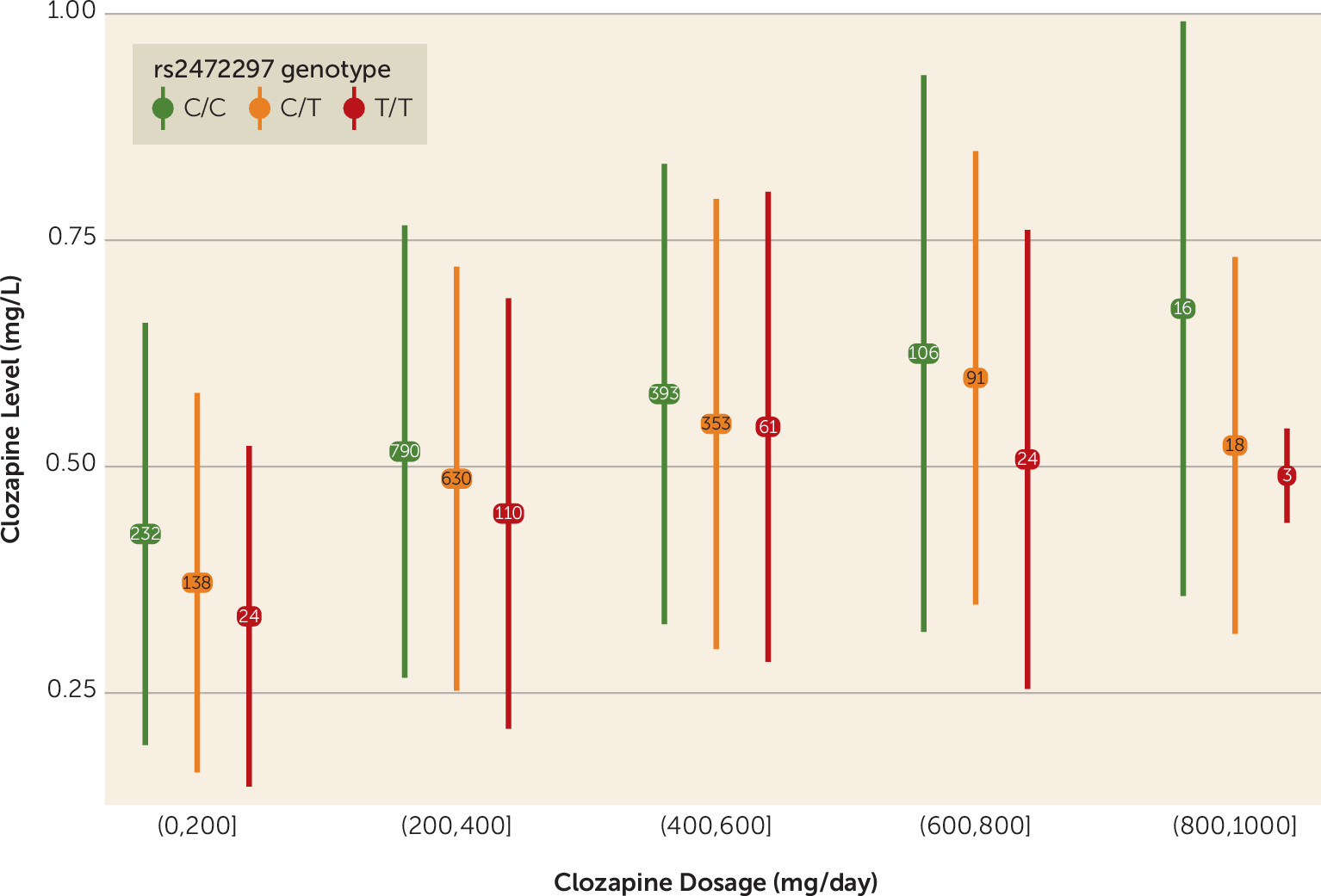
CYP1A1/CYP1A2 is associated with a reduction in clozapine plasma concentrations roughly equivalent to a decrease in clozapine by 50 mg/day, and homozygosity for the minor allele is equivalent to a reduction by 100 mg/day (
Figure 2). Similar effects were found for the missense SNPs associated with norclozapine levels (see Figure S8 in the
online supplement). The impacts on clozapine metabolite concentrations captured by these SNPs warrant their further study within the context of personalized drug therapy, given their potential clinically significant impact on dosing.
In following up the results of the GWAS, we sought genomic regions associated with clozapine metabolism that have also been identified as influencing metabolism of other compounds. We identified a strong relationship between the genetics of clozapine and caffeine metabolism, a finding with potential clinical relevance. A link between clozapine and caffeine metabolism was first proposed on the basis of findings indicating that the results of caffeine clearance tests, used as an index of CYP1A2 activity, correlate with clozapine clearance (
59). While there have not been large-scale studies in clinical settings, the available evidence suggests that caffeine interacts competitively with clozapine, causing heavy coffee drinkers to have higher baseline clozapine plasma levels (
60). Among factors that may have obscured this finding in previous research are the proven correlation between smoking and coffee consumption (
61) and the observation that even decaffeinated coffee may lower the activity of some hepatic enzymes (
62). In this regard, our analysis of metabolic-genetic association data showed commonalities, which hint at potential interactions, with several compounds related to coffee and caffeine. Remarkably, loci outside of the widely studied CYP1A2 region seem to jointly affect both coffee consumption habits and plasma concentration of clozapine metabolites, as we have shown using a polygenic score approach (see the Supplementary Methods section of the
online supplement). However, given that we did not have access to coffee or caffeine consumption data, we could not assess the degree to which caffeine may mediate or moderate the genetic associations with clozapine metabolite levels. Nonetheless, our results add to the existing evidence of the potential clinical importance of the interaction between the metabolic pathways for clozapine and caffeine.
Our data can also be interpreted in the light of the proposed mechanistic link between smoking tobacco and clozapine metabolism, which is thought to result from induction by tobacco of CYP1A2 activity, which in turn increases the first-pass metabolism of clozapine (
63). Current guidelines state that patients on clozapine need to be more closely monitored if they stop smoking, as their plasma levels can suddenly rise as the CYP1A2 induction fades. This effect, also seen with other medications, has been attributed to the effect of polycyclic hydrocarbons present in tobacco smoke, rather than a direct action of nicotine (
64). Thus, nonsmoke alternatives to tobacco, such as nicotine patches and e-cigarettes, are generally considered not likely to interact with clozapine treatment. However, we have shown that genetic variants in UGT enzymes, which are responsible for nicotine glucuronidation, also have a role in clozapine metabolism. Specifically, we have highlighted a missense polymorphism in UGT2B10, previously shown to result in impaired enzymatic function (
65), as a credible causal variant for influencing norclozapine plasma levels. This enzyme has also been shown to be a substrate of several antipsychotic drugs with structural properties similar to clozapine’s (
66). Given that nicotine is a specific high-affinity inhibitor of UGT2B10, our results support the possibility of nicotine-clozapine interactions in the glucuronidation excretion pathway (
26,
66), which should be investigated in more detail.
One of the limitations of this study is that our regression models do not explain as much variance in plasma concentrations as previous studies (
16,
17). However, we note that these studies included the clozapine/norclozapine metabolic ratio as a covariate of clozapine plasma concentrations. Given our data, and considering all fixed-effect covariates (see Table S2 in the
online supplement), this addition would have increased the variance explained by our mixed model from 19.28% to 32.34%, but at the cost of adding collinearity and hindering its interpretability. In any case, these models likely represent a lower bound of variance explained for clozapine plasma concentrations, given that we lacked individual measures of some known predictors of clozapine metabolism, including smoking and weight. We attempted to address this limitation in the discovery GWAS by using a novel application of PRSs as genetically informative proxies of these measures (see the Supplementary Methods section of the
online supplement). While this did not affect the results, we acknowledge that these are just markers of the exposures and do not capture their full effects. A further limitation is the lack of detailed individual-level data on concomitant medications that could interact with clozapine. Coprescription of such medications (e.g., carbamazepine and fluvoxamine) has been shown to be rare given their potential for clinically important interactions (
67), and hence it does not seem feasible that such coprescription could be an important source of bias in our findings. Furthermore, given that the absence of detailed individual-level exposure data is known to obscure the detection of genetic influences in metabolic enzymes (
68), our finding of detectable GWAS signals is reassuring. A final limitation is the potential that those who have had their clozapine levels taken may be an unrepresentative sample of all those taking clozapine, which would constitute a form of selection bias. In examining this issue, we did not detect differences between those with or without clozapine therapeutic drug monitoring assays in the distribution of age, sex, and several PRSs (schizophrenia, IQ, body mass index, smoking). Nonetheless, we cannot rule out other selection effects, and thus our findings should be interpreted as relevant to the population in which clozapine levels are monitored.
In summary, our analysis has allowed us to dissect the clozapine metabolic pathway using genetic and pharmacokinetic data. We have also demonstrated commonalities with the metabolism of other biological compounds, in particular nicotine and caffeine, that highlight relevant facets of metabolism and indicate potential interactions of clinical importance. Furthermore, our findings indicate avenues for next-stage clinical studies to determine the utility of pharmacogenomic testing as a complement to clozapine monitoring procedures, with the potential to have an impact on clinical care through improved titration and dosing and minimizing of adverse drug reactions.