Intellectual disability refers to a highly heterogeneous group of childhood-onset disorders characterized by below-average intellectual functioning (IQ <70) and significant limitations in adaptive functioning, which covers many everyday social and practical skills (
3). Intellectual disability has an estimated prevalence of 2%−3% in the population; severe disability has a population prevalence of 0.3%−0.5% (
4). Intellectual disability is often monogenic, but many different genes and types of mutations are implicated (
3). ADHD is a common comorbid disorder in children with intellectual disability (
5). Studies of children with mild and borderline intellectual disability have identified ADHD in 8%−39% of cases (
5). A recent study using Swedish birth registry data showed that nearly all of this comorbidity can be attributed to genetic factors (
6). Based on such phenotypic and genetic overlap, it has been hypothesized that intellectual disability and ADHD, and neurodevelopmental disorders more broadly, have an overlapping genetic etiology (
6).
In this study, we evaluated the genetic overlap between intellectual disability and ADHD in an attempt to identify novel ADHD candidate genes. We investigated whether genes affected by rare mutations in patients with intellectual disability also contribute to ADHD risk through common genetic variation. For this, we used the latest data freeze from the Psychiatric Genomics Consortium (N=19,210) for discovery and the Lundbeck Foundation Initiative for Integrative Psychiatric Research (iPSYCH) sample (N=37,076) for replication. To provide functional support for the newly identified ADHD candidates, we used
Drosophila melanogaster, a model that can facilitate characterization of the involved neural substrates. The role of dopaminergic neurotransmission is well established in ADHD (
1). In addition, circadian genes and circuits have been implicated, as ADHD often goes together with sleep disturbances, and abnormal circadian rhythms of melatonin secretion have been observed in children and adult patients with ADHD (
7). Notably, positive genetic correlations between insomnia and sleep-related traits and ADHD exist (
8). Moreover, disrupting the activity of the circadian clock gene
Per1 in both mice and zebrafish revealed ADHD-like symptoms (
9). In
Drosophila,
Per1 mutants were found to be deficient for experience-dependent increases in sleep (
10). We therefore set out to investigate potential dopaminergic and circadian rhythm components of the identified phenotypes in
Drosophila. Dissecting the role of neuronal circuits can help pinpoint the neurotransmitter systems contributing to ADHD as a first step toward individualization of treatment. On down-regulation of gene expression pan-neuronally and in relevant neuronal subsets, we assessed locomotor activity, sleep, and related parameters as behavioral readouts, which we previously established to be relevant for ADHD (
11).
Discussion
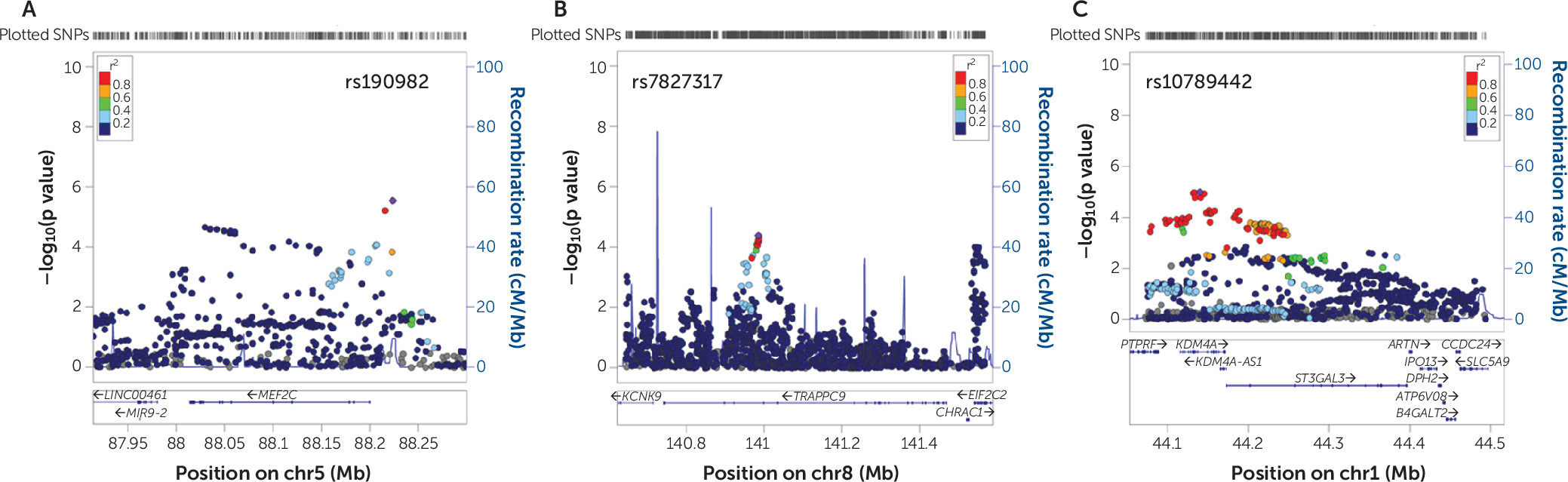
In this study, we used a robust discovery-replication design in the currently largest available independent data sets to show that genes affected by rare genetic variation in patients with intellectual disability also contribute to ADHD risk through common genetic variation. In the discovery phase, we also used different algorithms to test gene set association to further test the robustness of findings. In the KGG HYST test and MAGMA, we found significance in both self-contained tests but not the competitive test. A nonsignificant competitive p value in the competitive test should be interpreted as an inability to disentangle the part of the polygenicity attributable to the genes in the gene set from the polygenicity “remaining” (i.e., not captured by the set) on the rest of the genome. In combination with a significance in the self-contained test, it should not be interpreted as no effect of the selected gene set on the outcome. Our replication in the larger independent data set makes this point convincingly. Even more convincing is the fact that two of the three novel ADHD candidate genes that we identified,
MEF2C and
ST3GAL3, are among the genome-wide significant findings in the recently published ADHD GWAS-MA (
2).
Interestingly, our study design produced reproducible findings in much smaller sample sizes than those needed to reach genome-wide significance, which makes such overlap studies an attractive source of genes that have not previously been implicated by GWAS in ADHD. While we based our selection of intellectual disability genes on a diagnostic gene panel, many more intellectual disability genes are currently being discovered through the rapid advances in next-generation sequencing technology; those surely leave additional ADHD genes to be identified.
Our interdisciplinary approach, combining highly powered statistical analyses in humans with functional analyses in an unconventional, validated
Drosophila model for ADHD-related behavior (
11,
27) allowed for a direct validation and further characterization of neural substrates involved. None of our top three genes had been investigated in the context of ADHD before.
MEF2C encodes a member of the MADS box transcription factor, which binds to the conserved MADS box sequence motif (
28).
MEF2C is important for normal neuronal function by regulating neuronal proliferation, differentiation, survival, and synapse development (
29,
30). It also plays a role in hippocampal-dependent learning and memory, possibly by controlling the number of excitatory synapses (
31). While both haploinsufficiency and gene duplications of
MEF2C give rise to intellectual disability in humans, most severe cases of intellectual disability are linked to large deletions removing part or all of
MEF2C and de novo point mutations in the gene (
32); individuals with duplications of
MEF2C usually display a milder phenotype, with only mild cognitive impairment (
33). This is why we chose to model reduced gene expression in
Drosophila in this study. Common variants (SNPs) in the
MEF2C locus have previously been found to be associated with various cognitive, neuropsychiatric, and neurodegenerative phenotypes, such as intelligence (
34), schizophrenia (
35), and Alzheimer’s disease (
36), indicating pleiotropic effects of this gene on a range of phenotypes. The findings of our study add ADHD to this list and suggest that this is linked to the role of
MEF2C in neurotransmission, contributing to it through dopaminergic neurons. However, knowing that in
Drosophila dMEF2 expression is important in maintaining normal circadian rhythm (
37,
38), we cannot yet rule out an additional role of
dMEF2 in circadian neurons in the ADHD-related behaviors, as our
dMEF2 knockdown did not yield flies. This could be a result of
dMEF2 knockdown in circadian neurons causing cell death. Previous studies on circadian neuron ablation have indeed shown that this leads to embryonic lethality (
39). However, since
tim-Gal4 has been shown to be expressed outside the nervous system in the fat body and in oenocytes (
40,
41), we cannot exclude the possibility that
dMEF2 causes this lethality in noncircadian neurons.
In humans, four
MEF2 genes (A–D) exist, whereas in
Drosophila only a single gene represents the
MEF2 family (
dMEF2). In this study, we investigated the neuronal function of dMEF2 as a model of human MEF2C function and associated phenotypes, since we found that genetic variation in
MEF2C is primarily associated with intellectual disability, whereas genetic variation in
MEF2A is mainly linked to coronary artery disease and myocardial infarction, and no clinically relevant entries were found for
MEF2D (
42).
TRAPPC9 has been implicated in NF-kB signaling and is possibly involved in intracellular trafficking.
TRAPPC9 is highly expressed in postmitotic neurons of the cerebral cortex, and MRI analysis of affected patients showed defects in axonal connectivity (
43). The
Drosophila TRAPPC9 has been studied for its involvement in meiotic division in
Drosophila male gametes (
44), but a neuronal function has not been described so far.
TRAPPC9-associated intellectual disability is linked to loss of function of the gene (
45). Hyperactive behavior has so far been reported in one patient with a
TRAPPC9 mutation (
46). Our findings indicate that
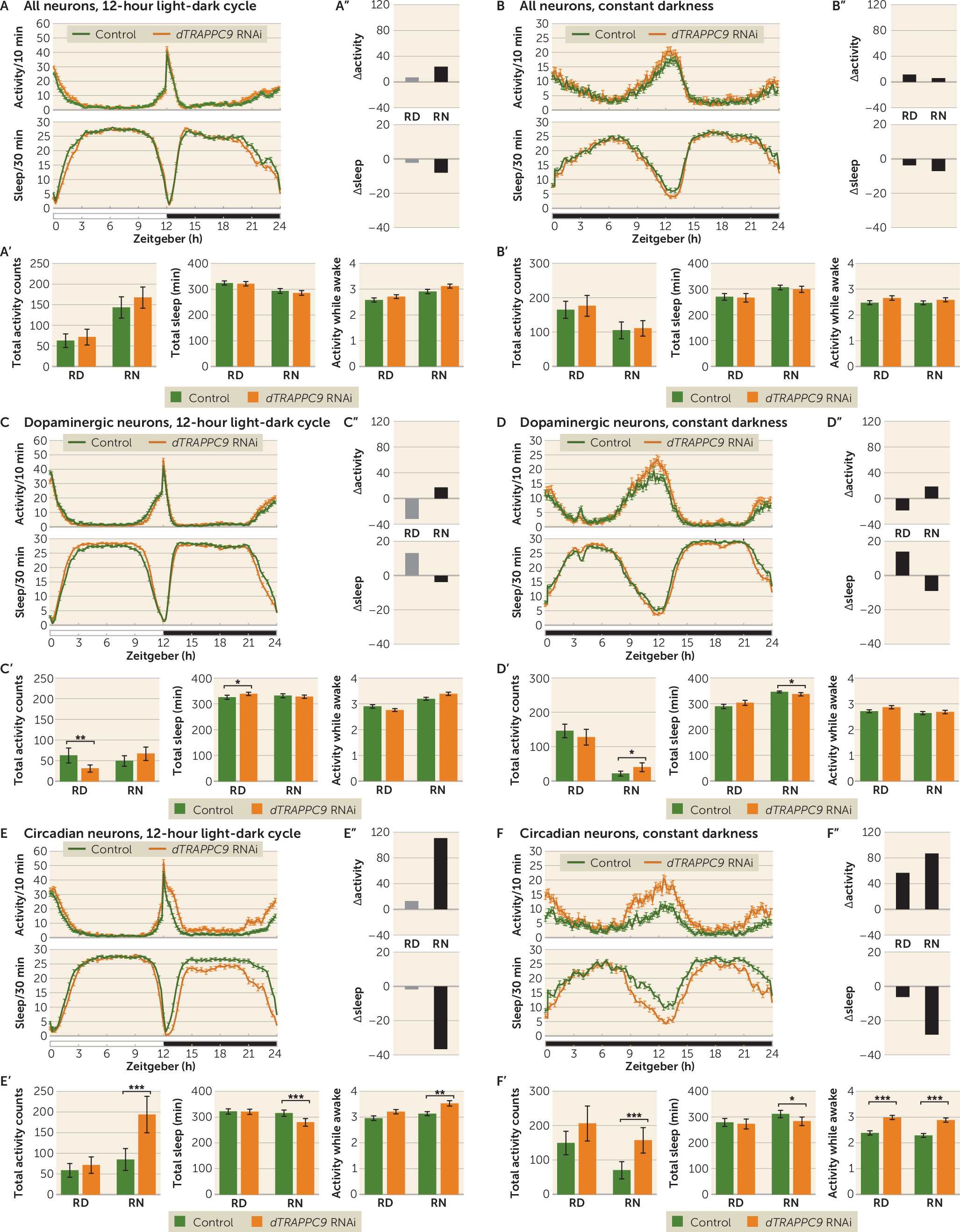
TRAPPC9 can play a role in ADHD and suggest that the gene acts primarily by affecting neurons involved in circadian regulation. Interestingly, while the
dTRAPPC9 dopaminergic neuron knockdown showed similar night activity and sleep profile to that of the control, somewhat lower activity and increased sleep were observed during the day, suggesting that
dTRAPPC9 has several cell-type-specific roles in the brain.
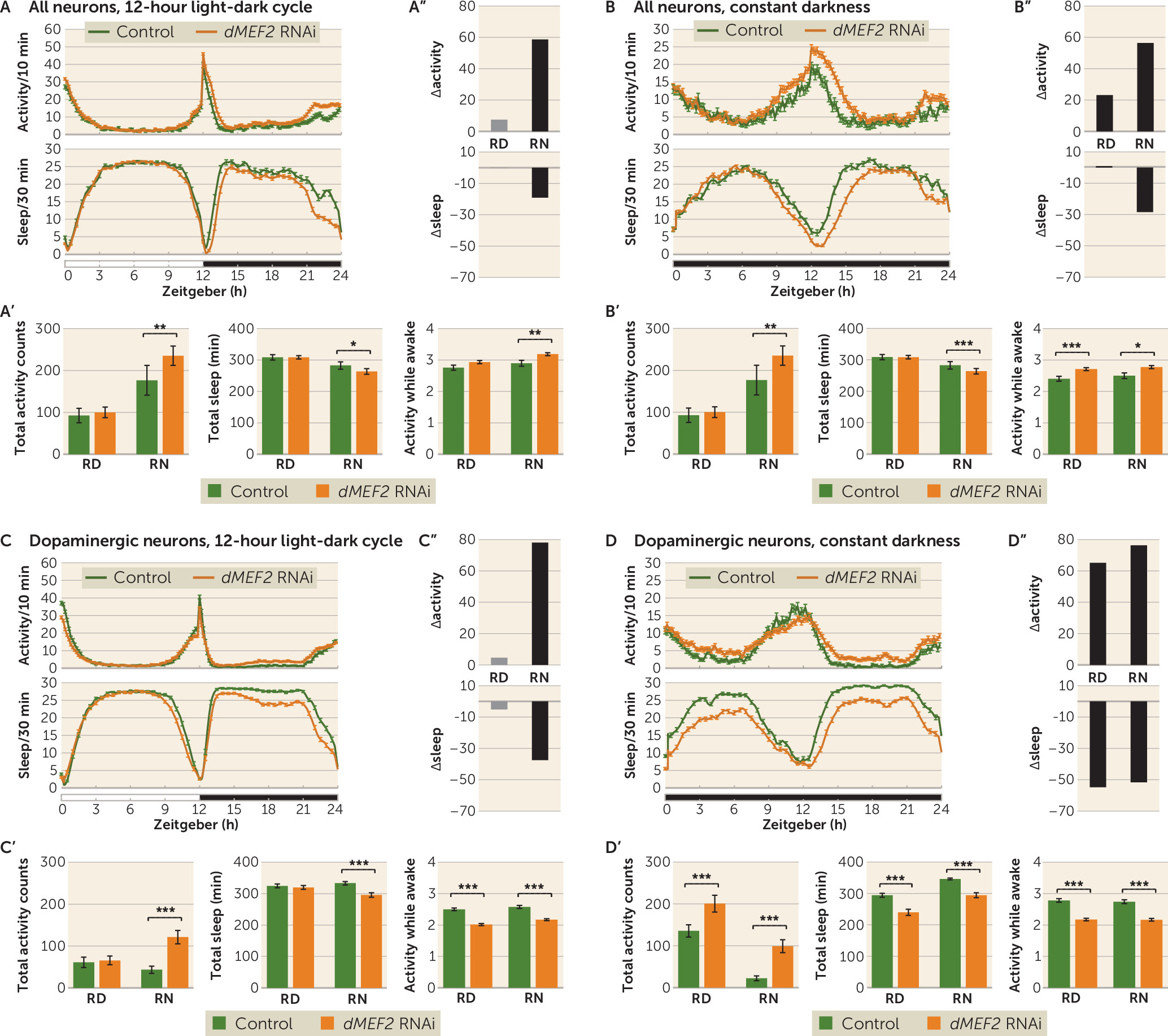
While both
dMEF2 and
dTRAPPC9 pan-neuronal knockdown showed weak or no activity nor sleep phenotype compared with the control, the knockdown in specific neuronal populations did result in pronounced alterations in activity and sleep profiles. Several explanations are possible for the observation of a more pronounced phenotype with specific neuronal subtype knockdown than with a pan-neuronal knockdown; the two most likely ones are 1) that a cell-type-specific driver is stronger in the specific neurons compared with a pan-neuronal driver, and 2) that a targeted gene functions in different circuits controlling the behavior of interest (e.g., excitatory versus inhibitory) that are differentially affected if a cell-type-specific knockdown is performed. Notably, in part of the
Drosophila brain, Sitaraman et al. (
25) identified distinct neuronal subtypes that oppositely regulate sleep. Considering that the whole nervous system is a mixture of various neuronal populations, each with a specific function in specified phases of development, the different activity and sleep profiles of
dMEF2 and
dTRAPPC9 knockdown in distinct neuronal populations indicates the need to investigate gene function in different brain circuits and identifies a particular strength of our study. A third possibility to explain the phenotypic differences between lines are differences in the developmental timing of knockdown between the genotypes, as determined by the expression of the utilized drivers. For the latter,
nSyb is known to be expressed earlier in development than
ple and
tim (
47); while this does not readily support timing of driver-determined expression as a cause of the different phenotypes, the possibility cannot be excluded. Importantly, our findings also show that different behavioral characteristics can contribute to ADHD-like activity phenotypes downstream of different gene defects; the
dMEF2 knockdown showed increased activity and sleep loss as a result of sleep defects, while in
dTRAPPC9 knockdown flies, the altered activity and sleep were caused by hyperactivity. Future studies will provide a more in-depth characterization of the neuronal morphology upon
dMEF2 and
dTRAPPC9 knockdown. Such studies are needed in order to better understand the impact that RNAi has on the morphology and functionality of the relevant neurons. There are several possible explanations for why the observed phenotype arises upon
dMEF2 and
dTRAPPC9 knockdown: loss of neurons, affected function of these neurons as a result of illness, or disruption of a more specific neuronal process. A similar phenotype was even reported to be caused by hyperactivation of dopaminergic neurotransmission (
48) and not by loss of dopaminergic neurons (
11). However, the ablation of circadian neurons does cause loss of circadian rhythm (
39,
49), which we did not observe upon
dTRAPPC9 knockdown. Although neuronal loss might still be a possibility, it is most likely that the phenotype is a result of cellular disturbances at the functional level.
The third ADHD candidate we identified,
ST3GAL3, is not conserved in
Drosophila, so we were not able to study its contribution to ADHD-relevant behavior. The gene encodes a membrane protein (ST3GalIII) that adds sialic acid to the terminal site of glycolipids or glycoproteins. The gene is expressed in a variety of tissues, including neurons (
50). In mice,
St3gal2 and
St3gal3 are responsible for nearly all the terminal sialylation of brain gangliosides and play an important role in cognition (
50). A role in brain development is also likely in humans, as the human brain is particularly enriched in sialic acid–containing glycolipids (i.e., gangliosides) (
51). Gangliosides are known to modulate calcium homeostasis and signal transduction in neurons (
52). Common genetic variants in
ST3GAL3 have also been associated with educational attainment (
53). Interestingly, in a recent study of DNA methylation, sites annotated to
ST3GAL3 were found to be associated with ADHD symptom trajectories in the population (
54). The use of alternative animal models, such as mouse or zebrafish, is warranted to characterize the neuronal circuits underlying
ST3GAL3’s effects on ADHD-related behavior.
In this study, we modeled one of the two behavioral symptom domains of ADHD, namely, hyperactivity. This was consistent with our findings—although only nominally significant, likely because of limited sample size—that
MEF2C and
TRAPPC9 were more strongly associated with hyperactivity/impulsivity than with inattention. However, being able to assess gene effects related to inattention would likely help to elucidate additional neural substrates and circuits involved in ADHD. Currently, multiple paradigms are available to assess attention in
Drosophila, as summarized by de Bivort and van Swinderen (
55).
In summary, the genetic overlap we observed between intellectual disability and ADHD may suggest biological pleiotropy, in which genetic variation severity in an overlapping set of genes is linked to the severity of neurodevelopmental phenotypes. Functional characterization of neural substrates involved revealed that the novel ADHD candidate genes may affect disease etiology through different biological pathways.