Early Detection and Preventive Intervention in Schizophrenia: From Fantasy to Reality
Abstract
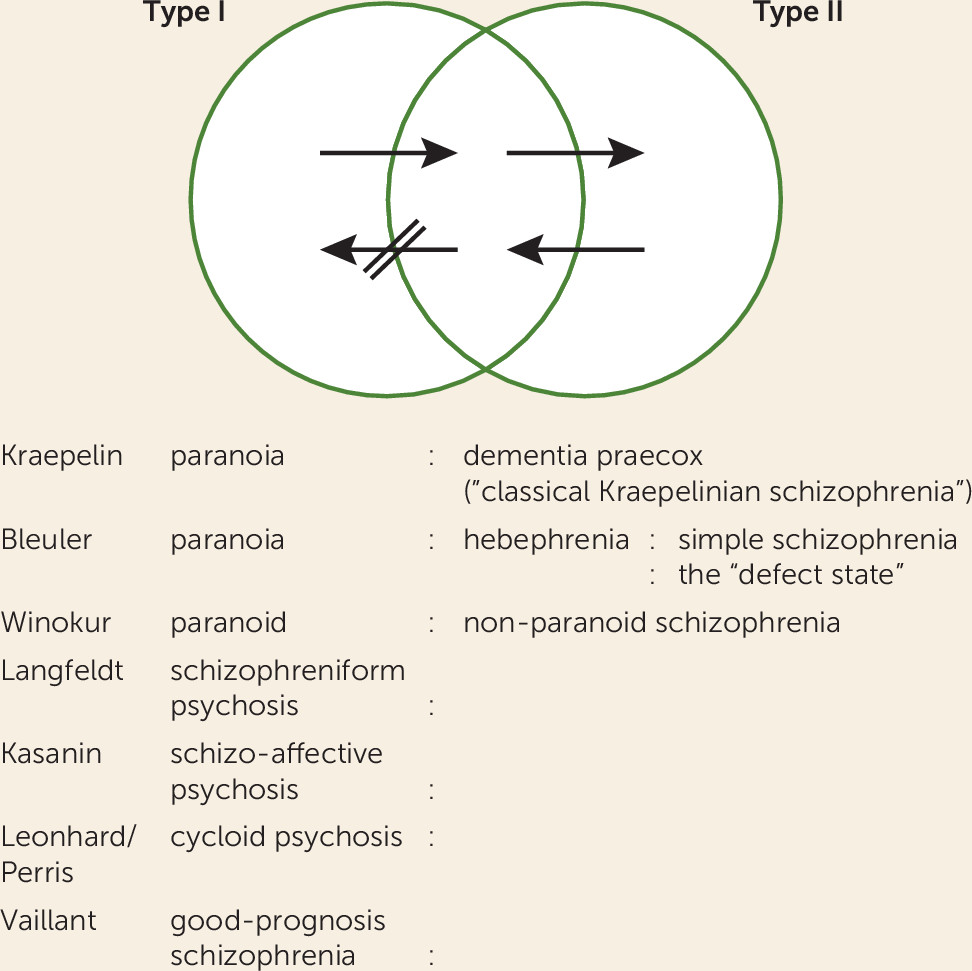
Nosology: the Early Classification of Schizophrenia
| Milestone | Authors, Year (Reference) | Significance |
|---|---|---|
| Defined dementia praecox | Pick 1891 (15), Kraepelin 1896 (16) | Established nosology of mental illness |
| Defined schizophrenia | Bleuler 1908 (18) | Redefined dementia praecox as schizophrenia |
| Published paper on prevention of onset | Sullivan 1927 (65) | First proposed idea for prevention |
| Discovery and early use of chlorpromazine | Laborit 1950 (cited in Thuillier 1999 [1]), Lehmann and Hanrahan 1954 (146), Delay and Denniker 1955 (147) | Development of antipsychotics and confirmation of effectiveness |
| Dopamine hypothesis | Snyder et al. 1974 (19), Seeman and Lee 1975 (148), Meltzer and Stahl 1976 (149) | First scientifically credible pathophysiological theory of schizophrenia |
| Early brain imaging studies | Huber 1957 (150), Haug 1982 (151), Johnstone et al. 1976 (152), Weinberger et al. 1979 (153) | First in vivo neuroimaging studies showing structural abnormalities |
| Synaptic pruning hypothesis | Feinberg 1982 (120), Keshavan et al. 1994 (123) | Heuristic pathophysiological theory of schizophrenia |
| Neurodevelopmental hypothesis | Weinberger 1987 (31), Murray et al. 1992 (32), Bloom 1993 (33) | Heuristic pathophysiological theories of schizophrenia |
| Revised dopamine hypothesis | Pycock et al. 1980 (24), Carlsson 1988 (25), Davis et al. 1991 (26), Lieberman et al. 1997 (27), Laruelle and Abi-Dargham 1999 (28), Kapur 2003 (29), Howes and Kapur 2009 (30) | Heuristic pathophysiological theories of schizophrenia |
| Two-syndrome hypothesis | Carpenter et al. 1973 (154), Crow et al. 1982 (34), Andreasen and Olsen 1982 (155) | Heuristic pathophysiological theories, positive and negative symptoms of schizophrenia, two-syndrome typology of schizophrenia, positive and negative syndromes of schizophrenia |
| Genetic high-risk studies | Gottesman and Shields 1966, 1967 (67, 68), Erlenmayer-Kimling and Cornblatt 1987 (69), Mednick et al. 1987 (70), Mirsky 1995 (71), Hodges et al. 1999 (72) | Long-term high-risk cohort studies to identify predictors of schizophrenia |
| First-episode psychosis studies | May et al. 1981 (36), Rabiner et al. 1986 (12), Nuechterlein et al. 1992 (156), Kirch et al 1992 (7), Lieberman et al. 1993 (14), Thara et al. 1994 (13), Melle et al. 2004, 2008 (48, 49), Norman et al. 2011 (157), Nordentoft et al. 2014 (56), Dixon et al. 2015 (50), Kane et al. 2016 (54) | Prospective studies of early stages of schizophrenia demonstrated good treatment response, reduction of duration of untreated psychosis, feasibility and efficacy of specialized treatment programs |
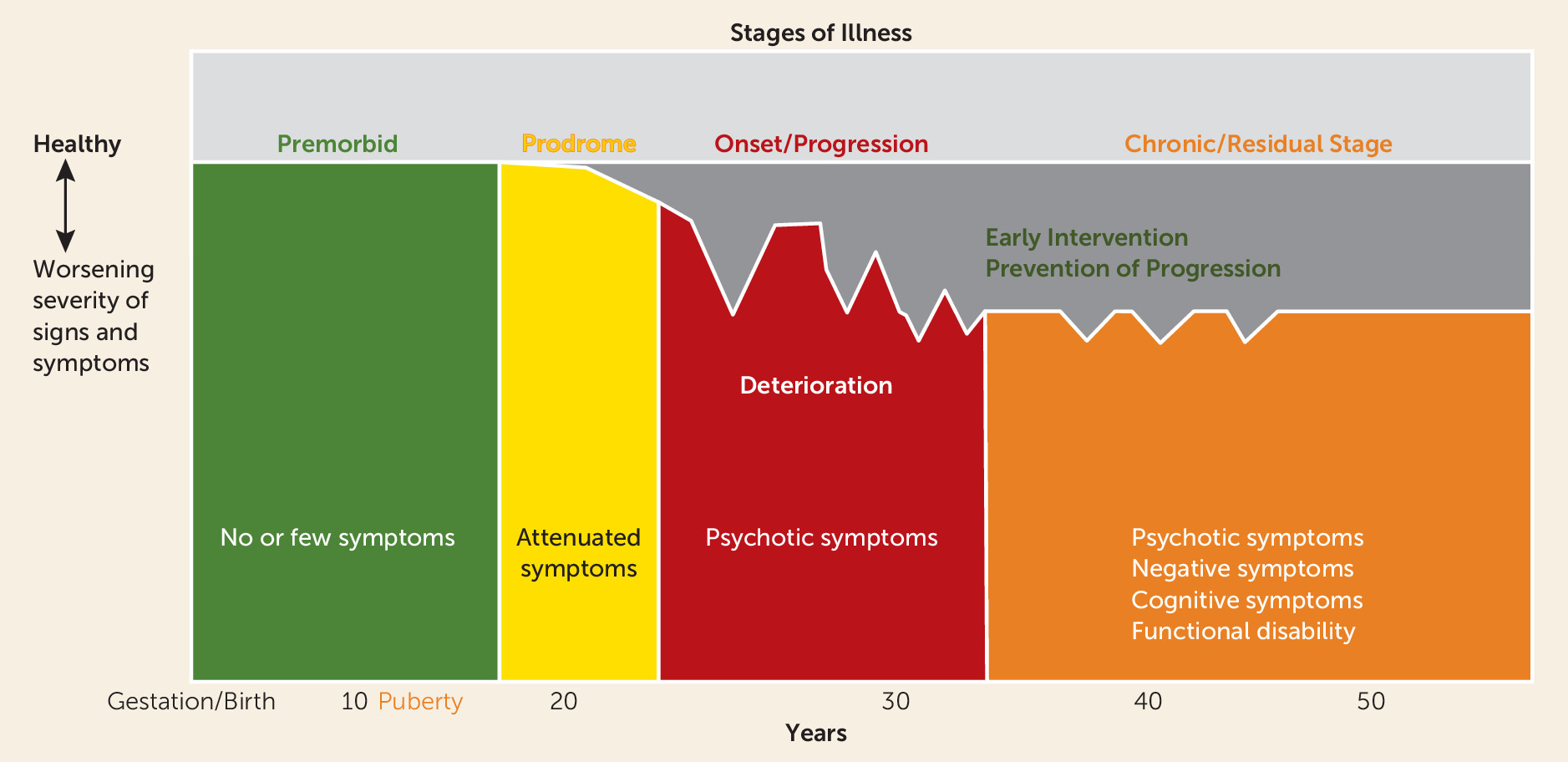
| Neuroprogression hypothesis of schizophrenia | McGlashan 1988 (8), Wyatt 1991 (9), DeLisi et al. 1997 (158), Lieberman 1999 (159) | Influential review of schizophrenia course found deterioration in early stages of illness inspired heuristic pathophysiological theory of schizophrenia and the early detection and intervention initiative |
| Glutamate hypothesis | Javitt and Zukin 1991 (128), Krystal et al. 1994, 2005 (137, 160), Olney and Farber 1995 (161), Goff and Coyle 2001 (162), Moghaddam and Javitt 2012 (129), Schobel et al. 2013 (93) | Heuristic pathophysiological theories of schizophrenia |
| Brain MRI studies of progression | Lieberman et al. 1992, 2001 (163, 164), DeLisi et al. 1997 (158), Gur et al. 1998 (165), Andreasen et al. 2011 (166), Cahn et al. 2002, 2006, 2009 (38–40), Bartzokis et al. 2012 (42), Schobel et al. 2013 (93) | Studies showing progressive brain morphologic changes over the course of schizophrenia |
| Clinical high-risk studies | Falloon 1992 (66), Yung and McGorry 1996 (74), McGlashan et al. 2001 (167), Addington et al. 2007 (77), Klosterkötter et al. 2005 (80), Ruhrmann et al. 2010 (81), Egerton, Howes et al. 2009, 2011, 2013 (98–100), Schobel et al. 2013 (93), McFarlane et al. 2015 (57), Brucato et al. 2017 (82), Koutsouleris et al. 2018 (78), Tognin et al. 2019 (79) | Clinical high-risk studies that demonstrated rates and time course of conversion and phenomenologic criteria for identification and diagnosis |
Efforts to Understand the Etiology and Pathophysiology of Schizophrenia
Studies of Natural History and Early Intervention
Duration of Untreated Psychosis and the TIPS Study
First-Episode Psychosis Studies and the RAISE Initiative
Pre-Syndromal Detection and Intervention Research
History
Diagnostic Criteria
Diagnostic Biomarkers
| Authors, Year (Reference) | Biomarker | Modality | Finding and Significance |
|---|---|---|---|
| Ferrarelli et al. 2007 (168), Manoach et al. 2016 (101) | Sleep spindles | High-density EEG | Sleep spindles are reduced in schizophrenia. They are generated by the thalamic reticular nucleus and dorsal thalamus, then transferred by thalamic relay neurons to the cortex, where spindle oscillations are synchronized. Current pathophysiologic models of schizophrenia highlight the possibility of thalamocortical dysfunctions being related to GABA and NMDA receptor-mediated glutamatergic neurotransmission accounting for core symptoms of illness. |
| Schobel et al. 2009, 2013 (93, 102) | Hippocampal CA1 cerebral blood flow and volume | MRI: structural and gadolinium | Elevated left CA1 cerebral blood volume at baseline predicts transition to syndromal psychosis and hippocampal atrophy. |
| Howes et al. 2009, 2011, Egerton et al. 2013 (98–100) | Dopamine synthesis capacity in striatum | PET: F-DOPA | Individuals at high risk for psychosis have elevated striatal dopamine, particularly in the associative striatum, and this predicts transition to syndromal psychosis. |
| Stone et al. 2009 (169) | Glutamate levels in anterior cingulate cortex and thalamus and gray matter volume | 1H-MR spectroscopy and structural MRI | Higher glutamine levels in anterior cingulate in high-risk individuals compared with control subjects, but lower glutamate and glutamine (Glx) levels in thalamus than controls; less gray matter volume in orbitofrontal cortex and ventral anterior cingulate cortex and more gray matter volume in left cerebellum and left occipital cortex in high-risk individuals than control subjects. |
| de la Fuente-Sandoval 2011, 2013, 2015 (103–105) | Glutamate levels in associative striatum | 1H-MR spectroscopy | Individuals at high risk for psychosis have elevated levels of glutamatergic metabolites in the associative striatum, and this predicts transition to syndromal psychosis. |
| Bodatsch et al. 2011, Perez et al. 2014 (106, 107) | Mismatch negativity | MMN/EEG | Mismatch negativity is decreased in high-risk individuals and predicts transition to syndromal psychosis |
| Walker et al. 2013 (108) | Salivary cortisol | Salivary cortisol | High-risk individuals have higher salivary cortisol than control subjects, higher cortisol correlates with greater psychopathology, and baseline cortisol predicted conversion to psychosis. |
| Bedi et al. 2015 (109) | Automated speech analysis and machine learning | Automated speech analysis and machine learning | Speech features of individuals at high risk for psychosis are able to predict who will transition to a syndromal psychosis. |
| Anticevic et al. 2015 (110) | Thalamic dysconnectivity | Resting-state MRI | Thalamic dysconnectivity is present in individuals at high risk for psychosis and is most pronounced in individuals who progress to syndromal psychosis. |
| Perkins et al. 2015 (111) | Blood-based biomarkers | Blood-based biomarkers | Measures of inflammation, oxidative stress, and the hypothalamic-pituitary axis may enhance risk prediction. |
| Chung et al. 2018 (112) | MRI-based neuroanatomical maturity | Machine learning and MRI | High-risk individuals have a greater deviance in their neuroanatomical maturity than do healthy control subjects, and this predicted transition to syndromal psychosis among younger individuals. |
| Cassidy et al. 2019 (113) | Neuromelanin sensitive MRI | MRI: neuromelanin | NM-MRI signal is positively correlated with severity of positive symptoms in both schizophrenia and in clinical high-risk individuals. |
| Bossong et al. 2019 (170) | Hippocampal glutamate levels | 1H-MR spectroscopy | High-risk individuals who transition to syndromal psychosis have higher hippocampal glutamate levels than high-risk individuals who do not transition. High-risk individuals who have poor functional outcomes have higher hippocampal glutamate levels than high-risk individuals who have good functional outcomes. |
Preventive Intervention Studies
| Study | Intervention | Sample Size (N) | Length of Follow-Up | Finding | Notes |
|---|---|---|---|---|---|
| McGorry et al. 2002 (171) | 6 months of risperidone plus CBT versus needs-based intervention | 59 | 12 months from baseline | Early advantages for conversion to syndromal psychosis did not persist | Single-blind |
| Morrison et al. 2004, 2007 (172, 173) | 6 months of cognitive therapy or treatment as usual | 58 | 12 months from baseline | Cognitive therapy was superior for transition to psychosis and positive symptoms | Advantages for transition to psychosis did not persist at 3 years |
| McGlashan et al. 2006 (174) | 12 months olanzapine versus placebo | 60 | 12 months from baseline | Olanzapine protected against conversion but nonsignificantly | Side effects were problematic and led to attrition |
| Amminger et al. 2010 (175) | 12 weeks of omega-3 polyunsaturated fatty acids or placebo | 81 | 40 weeks | Rates of conversion to syndromal psychosis were lower in the intervention group | |
| Yung et al. 2011, McGorry et al. 2013 (176, 177) | 12 months of cognitive therapy plus risperidone versus supportive therapy versus cognitive therapy | 115 | 12 months from baseline | No significant differences in rates of transition to syndromal psychosis | |
| Addington et al. 2011 (178) | 6 months of CBT or supportive therapy | 51 | Up to 18 months from baseline | No difference in transition rates or symptom change | |
| Bechdolf et al. 2012 (179) | 12 months of integrated psychological intervention (CBT, skills training, cognitive remediation, psychoeducation) or supportive counseling | 128 | 12–24 months from baseline | The integrated psychological intervention was superior to supportive counseling for transition to psychosis or subthreshold psychosis | Patients had “basic symptoms” |
| Van der Gaag et al. 2012 (180) | 6 months of CBT or treatment as usual | 201 | 18 months from baseline | CBT was superior for transition to psychosis and symptom reduction | |
| Morrison et al. 2012 (181) | 6 months of cognitive therapy or monitoring of mental status | 288 | 12–24 months from baseline | No difference in transition rates | |
| Woods et al. 2013 (182) | 12 weeks of glycine or placebo in the placebo-controlled trial; 8 weeks of just glycine in the open trial | 8 in placebo-controlled trial; 10 in open-label trial | Up to 24 weeks from baseline | Glycine treatment led to improvements in SIPS score at 8 weeks | Variable levels of follow-up and intervention after 8 and 12 weeks |
| O’Brien et al. 2014 (183) | Family-focused therapy (18 sessions of psychoeducation, communication training, and problem-solving training) or enhanced care treatment, which consisted of three sessions of psychoeducation | 66 | 6 months from baseline | Individuals in the intervention group demonstrated greater improvements on measures of communication and conflict | Intervention performed with high-risk patients and their parents or significant others |
| McFarlane et al. 2015 (57) | 24 months of family-aided assertive community treatment to early first-episode patients and higher-risk patients, and community care to a lower-risk group | 337 | 24 months from baseline | Family-aided assertive community treatment was superior to community care for positive symptoms | Involved both first-episode and high- and low-risk patients, and interventions were assigned based on risk level |
| Piskulic et al. 2015 (184) | 40 hours over 12 weeks of auditory processing cognitive remediation therapy versus computer games | 32 | Up to 9 months from baseline | Within-group cognitive improvements over time (up to 9 months) | |
| Kantrowitz et al. 2015 (185) | 16 weeks of d-serine or placebo | 35 | 16 weeks from baseline | d-Serine had a beneficial effect on total negative symptoms | Included high-risk patients with negative symptoms |
| Loewy et al. 2016 (186) | 40 hours of auditory processing–based exercises designed to target verbal learning and memory operations or computer games | 83 | 2 months from baseline (40 hours of the intervention) | Auditory processing–based exercises improved verbal memory to a greater degree than did computer games | |
| Stain et al. 2016 (187) | 6 months of CBT or nondirective reflective listening | 57 | 12 months from baseline | Individuals in the nondirective reflective listening group experienced greater decreases in distress associated with subclinical psychotic symptoms | |
| McGorry et al. 2017 (188) | 6 months of omega-3 polyunsaturated fatty acids and cognitive-behavioral case management or placebo and cognitive-behavioral case management | 304 | 6 months from baseline | No differences in rates of transition to syndromal psychosis | |
| Bhattacharyya et al. 2018 (189) | One dose of cannabidiol or placebo | 33 | Cannabidiol resulted in intermediate activation in striatal, medial temporal lobe, and midbrain areas of the brain compared with high-risk individuals who received placebo and healthy control subjects | Used BOLD fMRI |
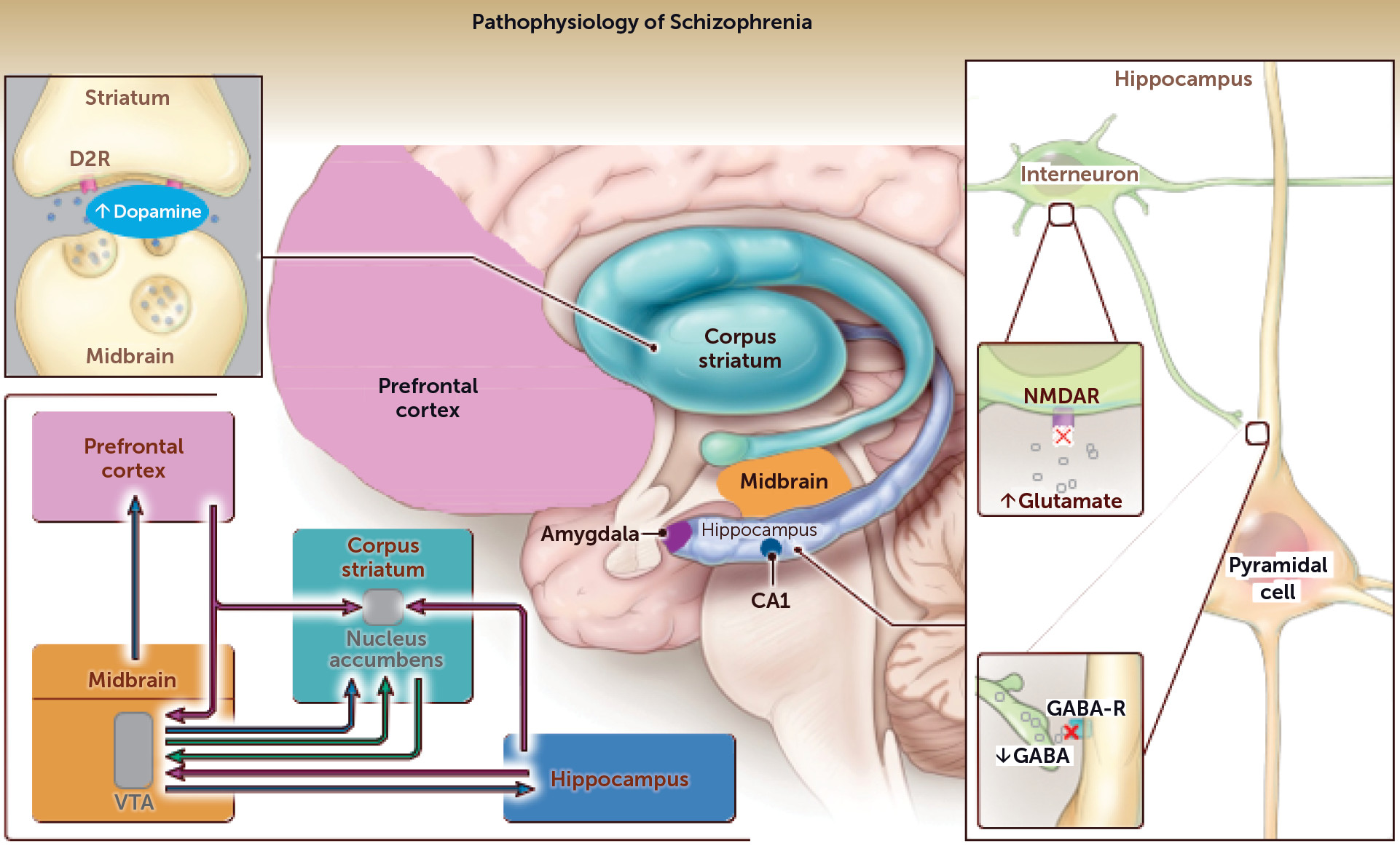
Pathophysiological Theories to Guide Future Research
Dopamine Theory
Synaptic Pruning/Immune Theory
Glutamate and GABA Theory
A Unified Model
Conclusions
Acknowledgments
Footnote
References
Information & Authors
Information
Published In


History
Keywords
Authors
Funding Information
Metrics & Citations
Metrics
Citations
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
For more information or tips please see 'Downloading to a citation manager' in the Help menu.
View Options
View options
PDF/EPUB
View PDF/EPUBLogin options
Already a subscriber? Access your subscription through your login credentials or your institution for full access to this article.
Personal login Institutional Login Open Athens loginNot a subscriber?
PsychiatryOnline subscription options offer access to the DSM-5-TR® library, books, journals, CME, and patient resources. This all-in-one virtual library provides psychiatrists and mental health professionals with key resources for diagnosis, treatment, research, and professional development.
Need more help? PsychiatryOnline Customer Service may be reached by emailing [email protected] or by calling 800-368-5777 (in the U.S.) or 703-907-7322 (outside the U.S.).