Death by suicide is a behavioral event that reflects a complex, heritable phenotype with diverse clinical antecedents and environmental risk factors (
1). The estimated annual age-adjusted prevalence rate of suicide death is approximately 0.014% in the United States (
2), and this rate has been steadily increasing since 2000 (
3). Suicide is now ranked the second leading cause of death among all persons 15–24 years old in the United States (
4). As such, suicide is considered a major public health challenge (
5), which has spurred research into its etiology and clinical prediction (
6,
7).
While it is established that suicide is significantly heritable (
8), well-powered genomic research on the topic has been largely limited to the study of suicide-related behaviors rather than the ultimate phenotype of suicide death. And although results from well-powered genome-wide association studies (GWASs) of suicidal behavior have advanced our understanding of the process (
9), the vast majority of suicidal behavior does not result in suicide death (
1,
10). Thus, suicidal behavior represents a less severe and likely more heterogeneous phenotype than suicide death, characteristics that may adversely affect statistical power to detect genetic associations. Conversely, the unambiguous phenotype of suicide death avoids several confounders inherent in the study of suicidal behavior or ideation and also focuses study on one of the single most critical contemporary public health outcomes. Previous genomic studies of suicidal behavior have also been limited by narrow ascertainment, examining only individuals with specific diagnoses (e.g., mood disorders, psychotic disorders) in order to maximize severity and accommodate post hoc study designs. Here, we avoided this limitation, instead using population-based sampling with ascertainment wholly independent of any co-occurring diagnoses, thus improving the representativeness of cases to the corresponding population and, consequently, the generalizability of results.
Suicide death, like its associated clinical antecedents, including schizophrenia and depression, has a highly multifactorial, and likely highly polygenic, etiology (
9,
11). Currently, the scientific literature lacks well-powered studies of suicide death in relation to molecular genetic risk for any medical or psychiatric diagnoses, and no robust polygenic scores have yet been developed for the critical outcome of suicide death. There are, however, a few modestly powered GWASs of suicide death in the literature (
12,
13), the largest of which combined two independent Japanese cohorts totaling 746 deaths by suicide (
13). Additionally, a recent UK Biobank GWAS of suicidal behavior conducted preliminary polygenic analyses of suicide death using a very modest number of deaths by suicide (N=127) (
14). Of note, these smaller studies reported evidence of polygenicity in both suicidal behavior and suicide death but with widely varying single-nucleotide polymorphism (SNP)-based heritability estimates, from an h
2SNP of 4.6 (
15) and 7.6% (
14) for suicidal behavior not including death to an h
2SNP of 35%−48% for suicide death (
13). The present study marks a notable advance from earlier research, because we used the world’s largest DNA data bank of suicide death, merged with a massive bank of electronic medical record and sociodemographic data (
16,
17), to model common variant genetic, and clinical phenotypic, precursors of suicide death.
This study adds to the growing body of genomic research on suicide (
13,
14), providing the first adequately powered GWAS of suicide death. In our analyses, we integrated data on modes of suicide death, medical and psychiatric diagnostic (ICD-10) codes (
18), and medical and psychiatric polygenic scores to comprehensively model common variant genetic risk for suicide death. Secondary analyses of sex differences were conducted as a result of the substantial sex differences in suicide death rates and modes of suicide death (
19,
20). Reliable and significant prediction of case-control status was achieved, adjusting for ancestry, using both a novel polygenic score for suicide death and polygenic scores for a range of comorbid psychiatric and medical risk factors, particularly behavioral disinhibition, major depressive disorder, and depressive symptoms. Additionally, gene set enrichment, SNP heritability, and genetic correlations were examined in this unique, unpublished data resource. We conclude with a brief discussion of the relevance of our findings to the broader field and promising future research directions.
Discussion
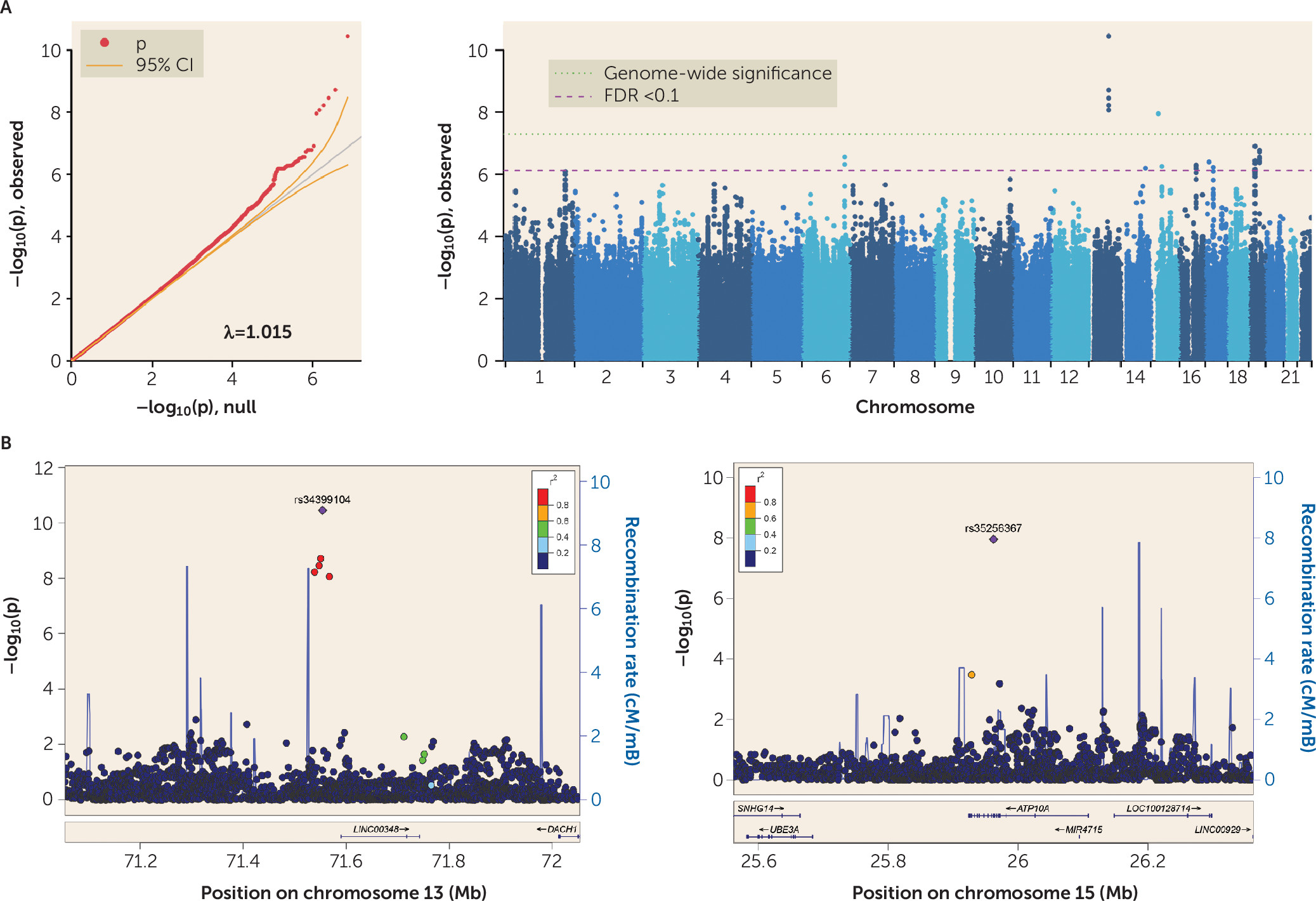
These results yield several insights and suggest important applications for future extension. To our knowledge, this is the first adequately powered comprehensive genomic study of suicide death. This GWAS of suicide death identified two genome-wide significant loci on chromosomes 13 and 15. The significant SNP-based heritability estimate of suicide death (25%, or 16% on the liability scale) is greater than that previously reported for suicide ideation and suicide attempt (5%) (
14,
15) and for suicide attempt within psychiatric diagnosis (3%−10%) (
43) and is closer to previous estimates reported for suicide death (35%−48%) (
13). Fully half of the genes implicated by our results overlap with schizophrenia results from the GWAS Catalog (p=1×10
−11), and two of these 11 genes (
HS3ST3B, NCAN) have previous associations with risk of suicidal behavior (for details on the relevant literature, see Table S25 in the
online supplement; for allele frequencies and effect sizes by cohort, see Table S26 in the
online supplement).
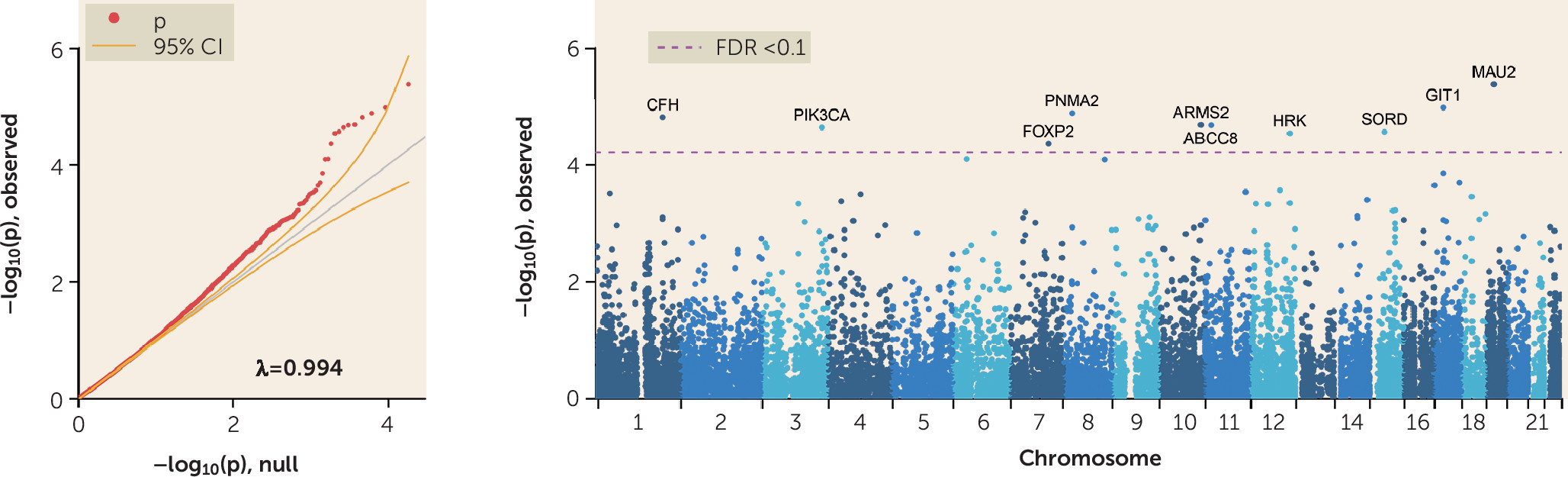
All but four of these schizophrenia-associated genes also drove a significant GWAS Catalog association with bipolar disorder (p=1×10–17), supporting the hypothesis of genetic liability to more nonspecific psychiatric illness. Of particular interest, the region on chromosome 19p13.11 in our study is supported by evidence from the most recent, well-powered studies of both bipolar disorder (
44) and schizophrenia (
45). In addition, our region on chromosome 16q21 is supported by evidence not only from several large GWASs of schizophrenia but also from a large meta-analysis of autism spectrum disorder (
45–
50).
Genome-wide significant SNPs on chromosome 13 (
Table 1) are all intergenic and vary with respect to a combined annotation-dependent depletion score, an index for scoring the deleteriousness of single-nucleotide variants as well as insertion and deletion variants in the human genome. Notably, the variant with the strongest GWAS effect size, single-nucleotide variant rs34399104, reaches a combined annotation-dependent depletion score of almost 20, indicating that it is in the top 1% of deleterious variants in the genome.
Direct comparison of the effect sizes in our GWAS with 110 effect sizes reported in previous studies (
12–
15,
43,
51) identified only one nominally significant variant in the largest GWAS of suicidal behavior (
43), rs4870888, nearest to
FER1L6 and
FER1l6-AS2. (See Table S27 in the
online supplement for a direct comparison of previously reported effect sizes with those in our study.) The single-nucleotide variant rs7989250 (chromosome 13) was reported for ordinal suicidality in the UK Biobank GWAS and reached a p-value threshold <0.05 for replication in the suicide GWAS conducted by Otsuka et al. (
13) but did not reach the threshold in our study (see Table S27 in the
online supplement).
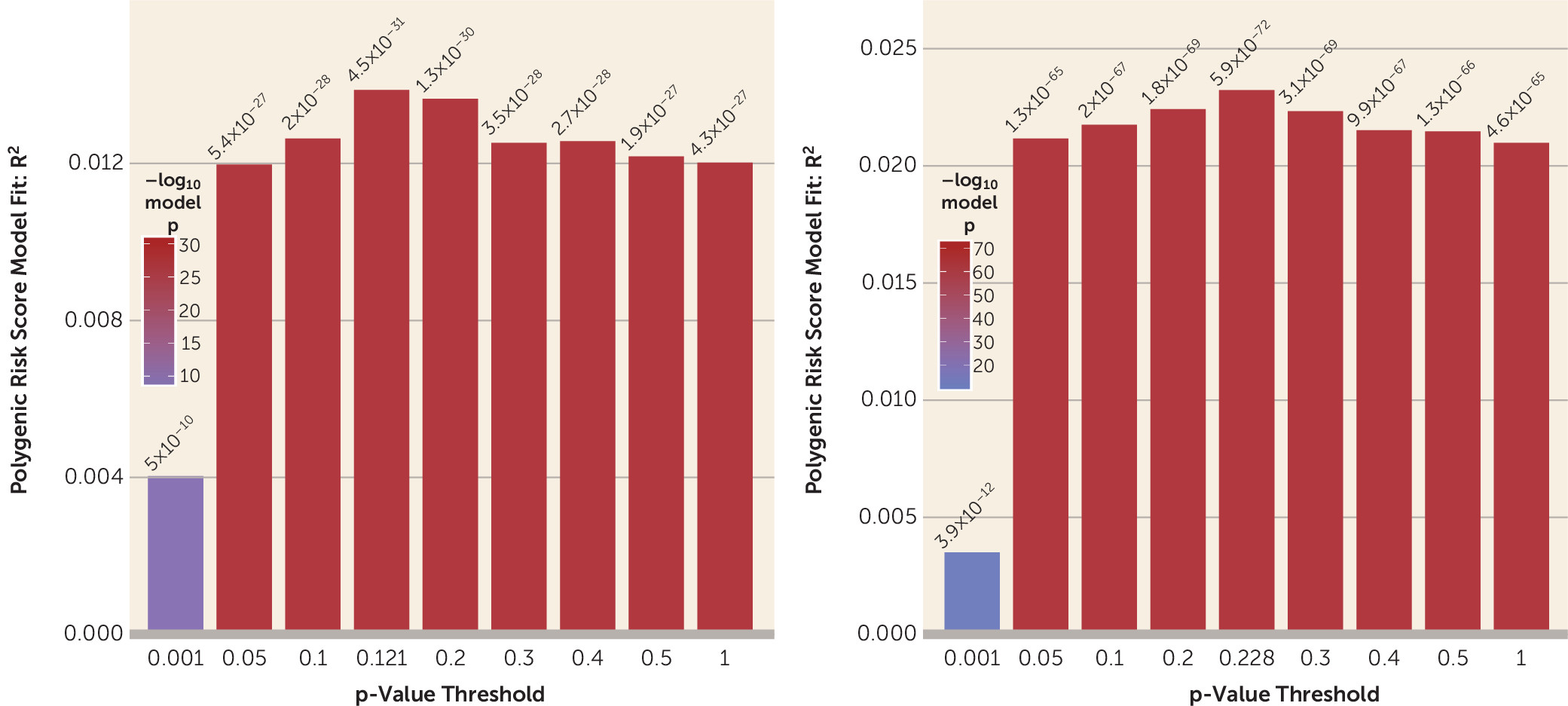
We were also able to cross-validate prediction of suicide death case-control status with polygenic scores for suicide death, accounting for population stratification. While within-case polygenic score validation relies on stronger assumptions than other polygenic score case-control comparisons, the considerable overlapping polygenic signal across p-value thresholds in this study (suicide deaths, N=3,413) is consistent with the two existing smaller polygenic analyses predicting suicidality (
13,
14). In one of those studies, suicidal behavior polygenic risk score predicted increased odds of suicide death, with 127 deaths (
14), and in the other, a significant SNP-based genetic correlation of suicide death was observed between two independent cohorts (suicide deaths, N=746) (
13).
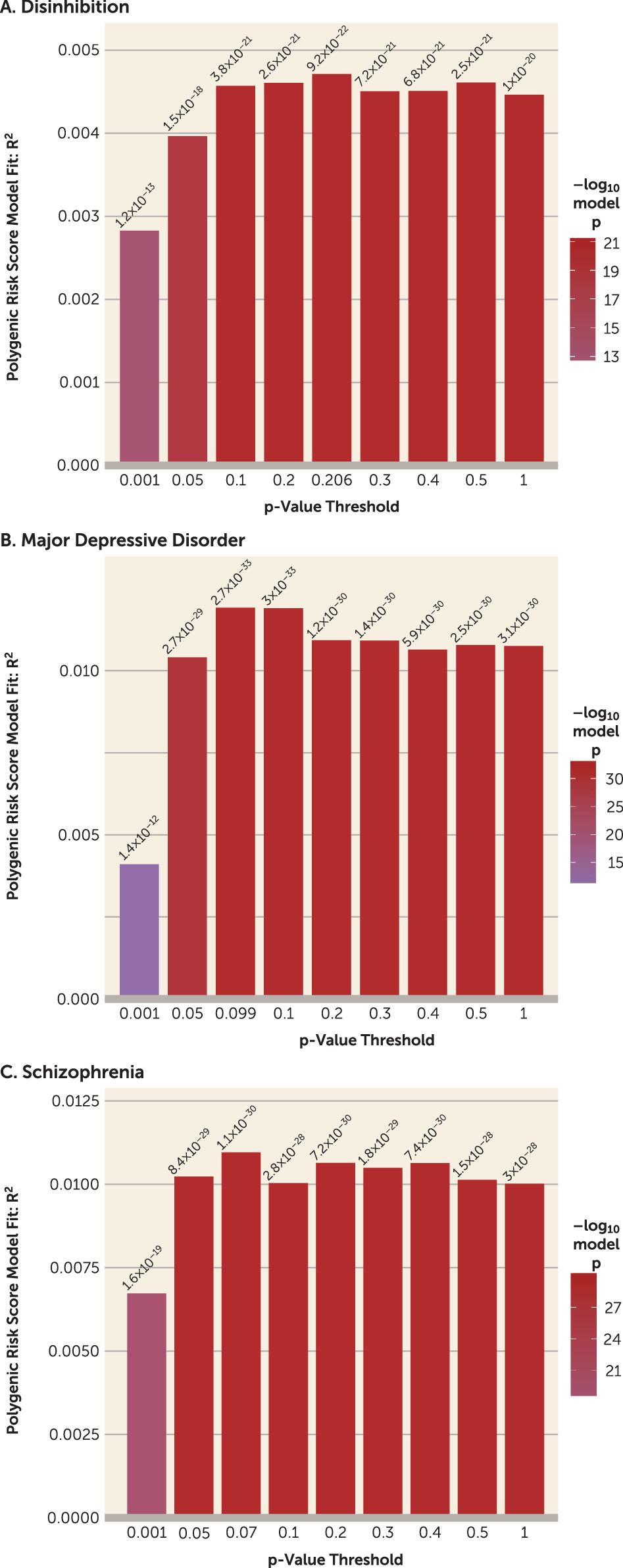
Perhaps most compellingly, suicide death was strongly associated with polygenic scores for multiple psychiatric and psychological traits. As noted above, these included alcohol use, autism spectrum disorder, child IQ, depression, disinhibition, schizophrenia, and loneliness (for additional polygenic risk score plots, see Figure S15 in the
online supplement). These results were consistent with expectations, because strong associations in case samples compared with both control cohorts were observed with behavioral disinhibition and major depressive disorder—arguably the two most critical antecedents of suicide death (
52). More generally, these results are consistent with the emerging consensus that genetic correlations across psychiatric diagnoses and traits are substantial (
53). Further research with an even larger suicide death cohort will be needed to determine the ultimate cause of these polygenic score associations with suicide death, with leading hypotheses arguing for the presence of a p-factor capturing general liability to psychopathology (
54), contrasting with those arguing for pleiotropic effects across a small set of psychiatric disorder clusters (
55).
Limitations
Several limitations to this study should be noted. First, it is possible that population stratification or platform confounders stemming from convenience control samples could lead to false positives. To address this risk, we employed very conservative filters of both loci and study subjects to minimize false positives. Values of λ
GC <1.05 are generally considered benign, although inflation in λ
GC is proportional to the sample size (
56). Control samples in both the Generation Scotland database and the UK10K Project cohort were extensively assessed for ancestral comparability to samples comprising individuals who died by suicide in both the 1,000 Genomes Utah CEU (
25) and Utah European ancestry databases, and both matched extremely well. Incorporating two separate control cohorts (and formal analytical comparison of these cohorts to the corresponding 1,000 Genomes populations) also allowed us to detect and filter out loci related to platform-specific technical artifacts and subtle variation in population structure before case-control analyses. Results of tests for differences in loadings for up to 200 principal components by cohort and by case-control status are presented in Table S28 in the
online supplement. Before and after imputation, we excluded any and all variants generating q values <0.1 in a control-control GWAS. Additionally, employing ancestral Mahalanobis (i.e., centroid) distance pruning, we have leveraged the fact that the CEU 1,000 Genomes Project subjects are largely ancestrally homogeneous with our case samples to restrict our control subjects (and case subjects) to a highly ancestrally homogeneous group.
It is also possible that the use of a mixed control group—both healthy subjects and those with common medical conditions, as in the UK10K Project—could have exerted a mild bias on effect estimates toward the null, which we do not view as highly problematic, given that the direction of the bias effectively reduces the risk of false positive findings. In the suicide case cohort, rates of common medical conditions are much higher than population base rates. However, in stratified control analyses, effect sizes specific to our case sample compared with those in the UK10K Project were elevated relative to the case sample from the Generation Scotland database, which is inconsistent with potential confounding of signal as a result of other disorders. Additionally, effect sizes for polygenic scores for other disorders, across 1,000 p-value thresholds, were far greater between case and control subjects than between control groups, where differences were negligible and distributions of polygenic scores often overlapped. None of the implicated variants were significantly different in allele frequency between control groups in the control-control GWAS. However, it should be noted that allele frequency was relatively low for the top variants across all cohorts.
Another limitation of this study lies in the use of electronic medical record data, in which prevalences for disorders are always confounded with age, and in a number of ways. For example, a missing diagnosis could be more or less likely in a given decade during which a case was assessed. Younger ages are more likely to have complete electronic medical record information yet are less likely to be diagnosed with comorbid medical conditions, schizophrenia, or personality disorders.
Importantly, there was insufficient diversity in the case cohort to examine effect sizes for any non-European ancestries. We expect a sufficient number of case samples for a Mexican-American ancestry GWAS within 5 years, and we will prioritize diverse ancestry groups in order to minimize potential health disparities stemming from homogeneous summary statistics (
57).
Future Directions
A recent review of suicide prediction models has indicated that positive predictive values for suicide attempt are quite high (0.9), but positive predictive values for suicide death continue to hover near zero (
58,
59). In this study, we have discovered multiple genome-wide significant loci and genes, strong polygenic signal, and significantly increased genetic risks for behavioral disinhibition, major depression, depressive symptoms, autism spectrum disorder, psychosis, and alcohol use disorder among case samples. Future modeling of multiple polygenic risks and phenotypic risk factors may help isolate important moderators of risk and improve objective risk measures of suicide death. Importantly, ethical challenges associated with predictive models of suicide death are significant and must be addressed proactively across psychiatric and medical domains (
6,
60).