Children exposed to severe, chronic stressors are vulnerable to a plethora of health problems across the lifespan (
1–
3). These problems span the continuum of what are traditionally understood to be mental (e.g., depression, posttraumatic stress, substance misuse) and physical (e.g., coronary heart disease, some cancers, autoimmune conditions) illnesses. Little is known about the behavioral and biological pathways underlying risk for this heterogeneous set of health problems.
To stimulate mechanistic research in this area, Nusslock and Miller proposed a neuroimmune network hypothesis (
4), which integrates an evolving body of preclinical and translational research (
5–
8). The framework begins with the observation that even under normal physiologic conditions, brain circuits involved in emotion regulation engage in bidirectional communication with peripheral immune cells that mediate inflammation (
5,
6). The framework goes on to hypothesize that severe chronic stress in childhood amplifies this crosstalk, initiating positive feedback loops between peripheral inflammatory activity and the brain’s developing cortico-amygdala and cortico-striatal circuits, which respectively mediate threat and reward processing. As a consequence of the enhanced crosstalk, stress-exposed children are hypothesized to display a phenotype consisting of chronic low-grade inflammatory activity, along with heightened threat responsivity and dampened reward processing. Over time, components of this phenotype are hypothesized to accelerate the pathogenesis of inflammation-mediated health problems.
There is evidence to support the constituent tenets of the neuroimmune network hypothesis. For example, studies have identified stress-related variations in brain development in children exposed to chronic stressors such as socioeconomic disadvantage and parental maltreatment (
9,
10). The networks that support threat and reward processing are especially sensitive to these stressors and display what appear to be durable changes in structure and function (
11). A separate literature has detailed the immunologic correlates of these stressors, demonstrating that exposed children display larger cytokine responses to microbial challenge and signs of chronic low-grade inflammation (
1,
2,
12). Based on initial findings in animals (
5,
7), studies have administered immune-activating stimuli to humans and found that the resulting inflammation affects functioning of neural circuits involved in threat and reward processing (
13–
15). Observational studies of humans have also consistently reported associations between activity of these brain circuits and steady-state levels of inflammatory biomarkers (
16).
Nevertheless, no published studies have examined the validity of the framework’s core hypothesis, namely, that children exposed to severe chronic stress will display the phenotype described above. Here, we sought to fill this gap by studying a sample of urban children from diverse socioeconomic backgrounds. Although children can experience a variety of chronic stressors, we focused on socioeconomic disadvantage here because it is highly prevalent in the United States. In 2017, 19% of American children resided in households with incomes below the federal poverty line. An additional 22% lived in households with incomes considered near-poor or low-income (
17). Although the experiences of these children vary considerably, on the whole, they are at increased risk for chronic stressors that include material hardship, neighborhood violence, family instability, harsh parenting, and repeated bereavement, among others (
18). Relative to their more advantaged peers, disadvantaged children also show indications of the dysregulation the framework specifies, including low-grade inflammation (
19,
20), higher neural threat reactivity (
21–
24), and blunted reward processing (
21,
25). Given this backdrop, we predicted that to the extent that children were facing socioeconomic disadvantage, they would display the phenotype, hypothesized in the framework, consisting of low-grade inflammatory activity coupled with higher amygdala responsivity to threat and lower striatal responsivity to reward.
Methods
Sample
The study involved 277 children from the Chicago area. To be eligible, they had to be in eighth grade (typically 13–14 years old), English-speaking, and in good health, defined as being nonpregnant, without a history of chronic medical or psychiatric illness, free of prescription medications for the past month, without acute infectious disease for 2 weeks, and without functional MRI (fMRI) contraindications. Each child gave written assent and a parent or guardian gave written consent. Northwestern University’s Institutional Review Board approved the protocol.
Because of venipuncture problems, inflammation data were missing for two children. Neuroimaging data were missing for a number of children (N=32 for the threat task; N=44 for the reward task), because they arrived too late to complete the tasks, were too obese or too anxious to enter the scanner, or had previously unrecognized structural anomalies. No usable data were available for another group of children (N=36 for threat task; N=59 for the reward task) because of technical problems with acquisition (brain outside field of view), excessive motion (>10% of volumes censored within a paradigm), or limited variability in behavioral response (for details, see the online supplement). Thus, the Ns were 207 for threat analyses and 172 for reward analyses. Children for whom fMRI data were missing were more likely to identify as Black (p=0.003; and 0.001 for threat and reward tasks). However, they were similar to the broader sample on age, sex, Hispanic ethnicity, pubertal status, and household income (p values, 0.11–0.80).
Socioeconomic Conditions
Each child’s parent or guardian completed an interview about family socioeconomic conditions. From these data, we computed household income-to-poverty ratio. For most federal programs in the United States, households with an income-to-poverty ratio <1.00 are considered to be living in poverty. In 2014, when the study began, the federal poverty threshold was $24,008 for a family of two adults and two children. To facilitate interpretation of the statistical models, we grouped children into four categories, following convention in the literature: those in poverty (≤0.99) and those whom we refer to as low-income (1.00–1.99), middle-income (2.00–3.99), and higher-income (≥4.00).
Low-Grade Inflammation
At the same visit, antecubital blood draw was performed. To minimize circadian variation, venipuncture was performed between 8:00 a.m. and 10:00 a.m. Children fasted for 8 hours beforehand to minimize dietary influences. We measured serum levels of five inflammatory biomarkers, chosen for their consistent relationships with early-life stress (
1,
2,
12): C-reactive protein (CRP), interleukin-6 (IL-6), interleukin-8 (IL-8), interleukin-10 (IL-10), and tumor necrosis factor-α (TNF-α). CRP was measured in duplicate by high-sensitivity immunoturbidimetric assay on a Roche/Hitachi cobas c502 analyzer. The cytokines were measured in triplicate with a four-plex immunoassay on an automated microfluidic platform (Simple Plex, ProteinSimple, San Jose, Calif.). Intra-assay coefficients of variation were 2.5%–5.1% (for additional details, see the
online supplement).
Most biomarkers were skewed and/or kurtotic, but we normalized their distributions with log10 transformations. We then standardized the logged values of each biomarker (mean=0; SD=1) and averaged the resulting z-scores to form a composite score (Cronbach’s alpha=0.63), where higher scores reflect more low-grade inflammation. (Readers may wonder about including IL-10 in the composite, since its functions are anti-inflammatory. However, IL-10 is only expressed under proinflammatory conditions, so it correlates positively with the other biomarkers assessed here.) The composite score has two advantages. Statistically, it reduces the number of tests performed—here, by 80%—and thus the rate of false positive results. Biologically, a composite better reflects in vivo conditions, where proinflammatory cytokines are released in a cascading fashion and have redundant and synergistic effects on target cells.
Threat and Reward Paradigms
At a separate visit, neural responsivity to threat and reward was measured using fMRI. Briefly, to capture amygdala threat responsivity, we administered a morphed faces task (
26), where children viewed images of actors displaying emotional facial expressions at varying intensities and were asked to indicate the gender associated with each face. Threat was operationalized as the amygdala’s reactivity to angry faces (relative to fixation). The amygdala response to neutral faces (relative to fixation) was used in specificity analyses. To assess reward, children performed a passive avoidance task (
27), comprising 96 trials. In each trial, a shape was presented. Children could respond with a button press, triggering one of four outcomes: win $50, win $10, lose $10, or lose $50. Or they could ignore the shape, in which case no monetary outcome transpired. Outcomes were probabilistic and pseudorandom; responding to two particular shapes triggered wins on 87.5% of trials, and responding to other shapes triggered losses on 87.5% of trials. Afterward, children were paid $5, regardless of performance. Reward response was operationalized as ventral striatal reactivity to winning money (relative to fixation). Ventral striatal reactivity (relative to fixation) to losing money was used in specificity analyses.
MRI Parameters, Preprocessing, and Analysis
Imaging data were collected using a Siemens Prisma 3-T scanner with a 64-channel phased-array head coil. Structural imaging consisted of a high-resolution navigated multiecho magnetization-prepared rapid acquisition gradient-echo sequence (TR=2300 ms; TE=1.86 ms, 3.78 ms; flip angle=7°; FOV=256×256 mm; matrix=320×320; slices=208; voxel size=0.8 mm
3). Functional images were acquired using a T
2*-weighted gradient echo planar imaging sequence (TR=2000 ms; TE=27 ms; FOV=240 mm; matrix=94×94; flip angle=90°). Data were analyzed according to standard procedures consistent with previous work (
27), using AFNI (
28). Blood-oxygen-level-dependent responses were extracted for each subject from anatomically defined masks, including an amygdala mask for the threat task (Eickhoff-Zilles Architectonic Atlas: 50% probability mask) (
29) and a ventral striatum mask for the reward task (accumbens-area map) (
30). (Further details of scanning and analysis are provided in the
online supplement.)
Statistical Analysis
To evaluate hypotheses, we estimated linear regression equations, using model 1 of PROCESS v3.4 (
31) in SPSS. The outcome was the inflammation composite score, and covariates included age, sex, self-identified racial (non-White=0; White=1) and ethnic (non-Hispanic=0; Hispanic=1) category, and pubertal status (self-reported on the Pubertal Development Scale [
32]). Predictors included covariates, a multicategorical variable reflecting income-to-poverty ratio, a mean-centered variable reflecting amygdala response to angry faces or striatal response to monetary reward, and a product term representing the interaction between income-to-poverty ratio and amygdala or striatal reactivity. The neuroimmune network hypothesis stipulates that in such a model, a statistical interaction should emerge whereby socioeconomic disadvantage increases the likelihood of children displaying a phenotype consisting of low-grade inflammatory activity, heightened threat responsivity, and dampened reward processing. All reported p values are based on two-tailed tests.
Discussion
To explain the health consequences of childhood adversity, the neuroimmune network hypothesis postulates that early stress amplifies crosstalk between peripheral inflammatory cells and networked brain regions involved in threat and reward processing (
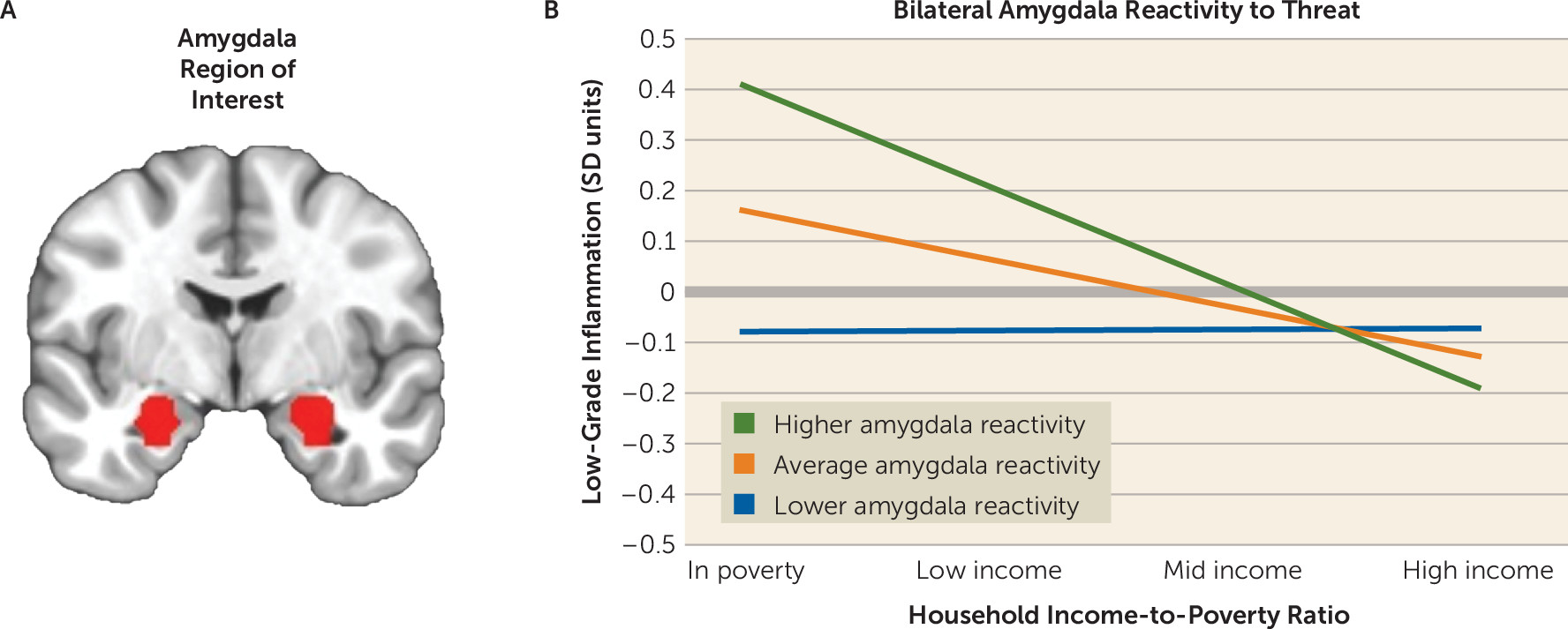
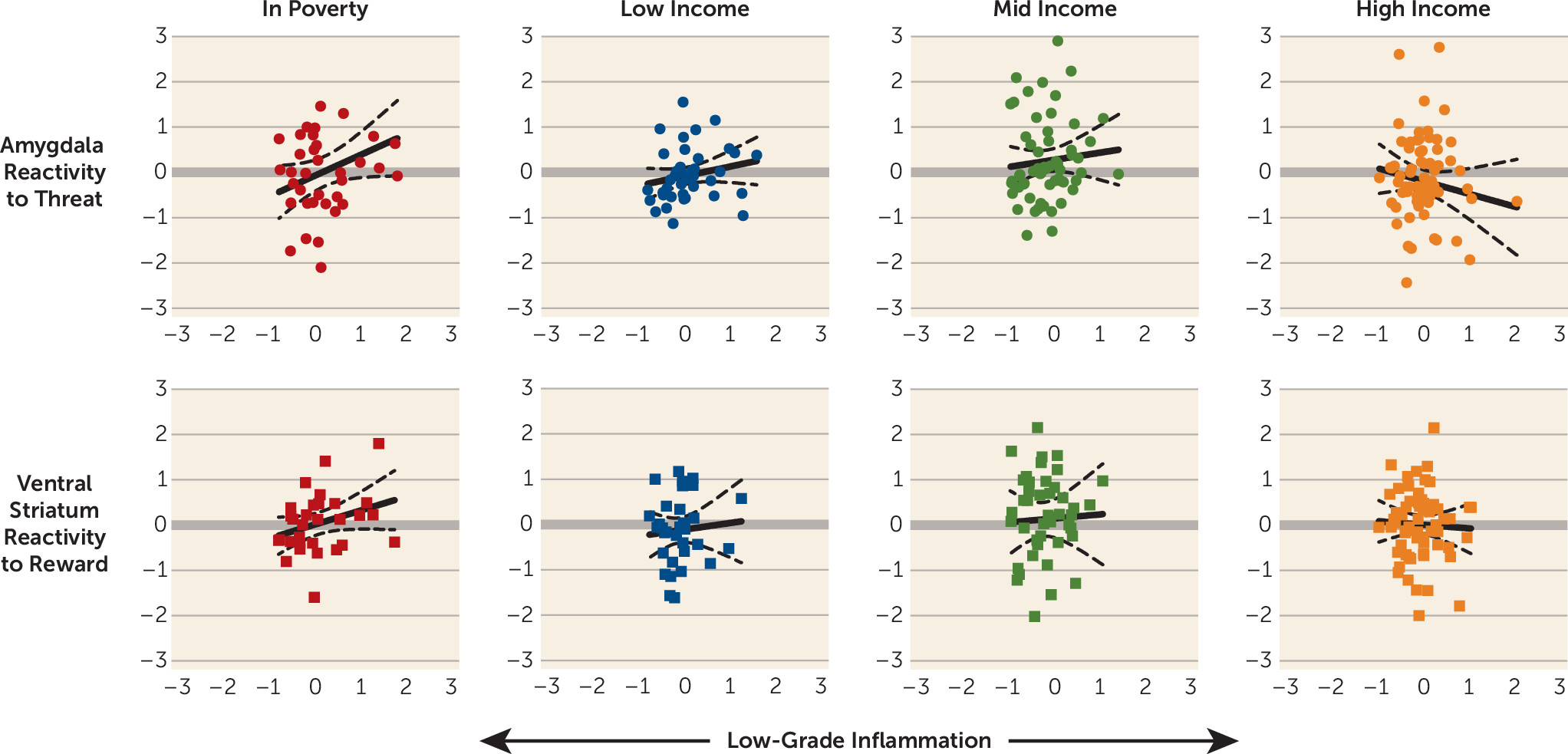
4). Although individual components of this hypothesis have been established in the literature, no published studies to date have integrated the data sources necessary to evaluate its core prediction. Here we sought to fill that gap in knowledge, using a diverse sample of urban children. In line with the core predictions of the framework, amygdala threat responsivity was positively associated with low-grade inflammation among children living in poverty. Intriguingly, the same pattern was evident for striatal reward responsivity. In households with more favorable socioeconomic conditions, these brain-inflammation associations were smaller in magnitude. The observed relationships were independent of demographic confounders and were robust to alternative measures of socioeconomic status. They also were condition specific: inflammation was associated with amygdala responsivity to threat stimuli, even when adjusting for activity during neutral trials, and with striatal responsivity to monetary rewards, even when adjusting for activity during losses.
The findings related to amygdala threat reactivity converge with previous evidence. Studies indicate that childhood disadvantage is associated with exaggerated behavioral, cardiovascular, and amygdala responses to threatening stimuli, a pattern thought to be adaptive in contexts where stressors can arise suddenly and unpredictably (
22,
34). Disadvantaged youths also exhibit indications of a proinflammatory phenotype, as reflected in higher circulating inflammatory biomarkers, and leukocytes that mount larger cytokine responses to microbial challenge and are relatively insensitive to inhibition by glucocorticoids (
20,
35). Our study’s findings bridge these distinct literatures and extend them by showing that the relationship between amygdala reactivity and inflammatory biomarkers is strongest among children living in poverty. Assuming this association reflects the crosstalk envisioned in the neuroimmune network hypothesis, it might help explain the excess health risks among disadvantaged children (
3). However, because of the study’s observational design, we cannot determine whether these associations reflect causal effects. With that said, experimental studies have illustrated the plausibility of causal effects, revealing bidirectional signaling between the amygdala threat circuitry and peripheral inflammatory cells (
14,
36). Such crosstalk is thought to be the substrate of an integrated network that detects threats involving microbial invasion and tissue damage, and subsequently mobilizes behavioral, physiologic, and immunologic resources for coping (
37).
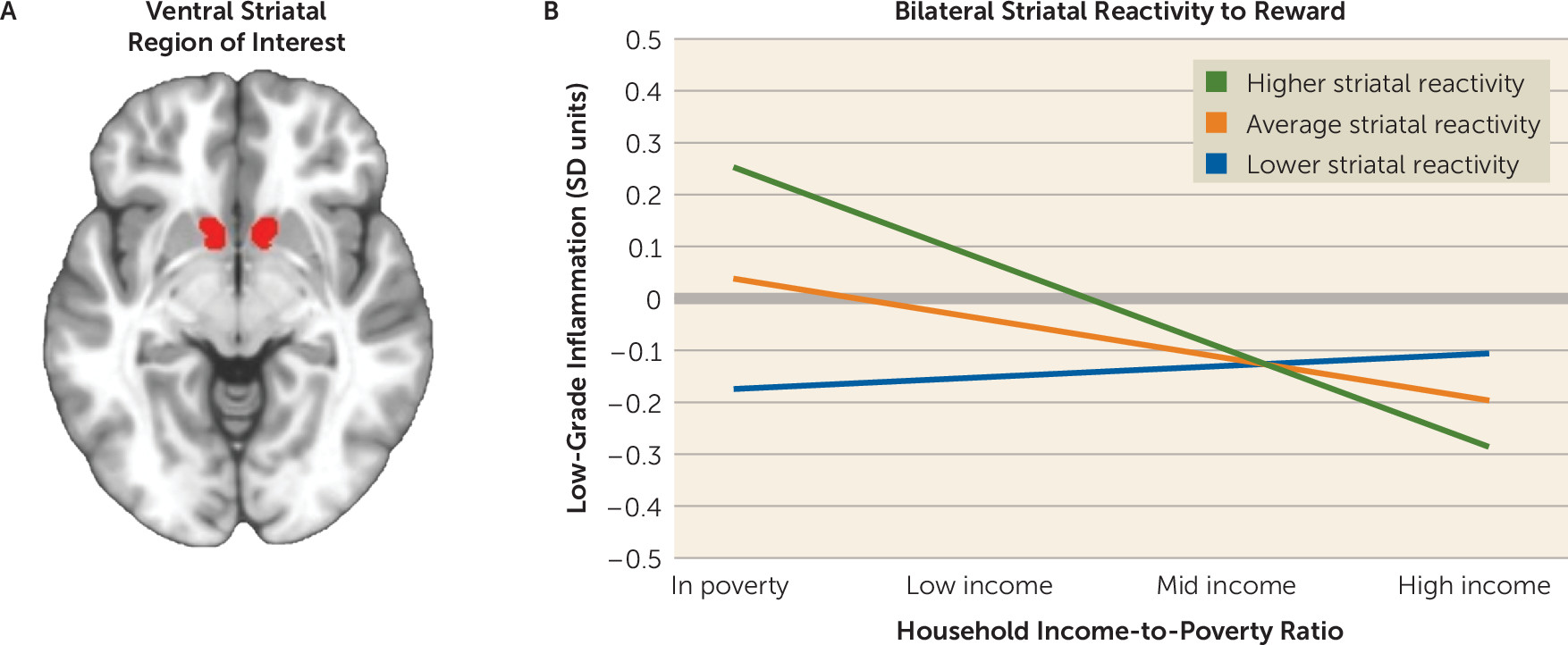
The findings involving reward responsivity in the ventral striatum were more complex. As predicted, children living in poverty displayed the strongest relationship between striatal responsivity and inflammatory activity. However, the direction of this relationship was positive, a pattern the original framework had not anticipated. With that said, the framework was formulated on the basis of studies from a decade ago, which showed that reward sensitivity dampens after administration of inflammatory agents (
13,
38). More recent studies indicate that this formulation is overly simplistic, as inflammation’s effects on striatal reward responsivity are context dependent (
8,
13,
39,
40). In situations where a reward matches what is motivationally salient to an individual, inflammation heightens striatal responsivity to that reward—for example, receiving social assistance when one is ill. In situations where there is a mismatch between a potential reward and what is motivationally salient, inflammation has the opposite effect, dampening striatal responsivity. This context dependence could explain the patterns seen here, because monetary rewards would be a highly salient reward for children living in poverty. Indeed, studies indicate that disadvantaged children display higher sensitivity to many appetitive stimuli, as reflected in temporal discounting, decision making, and self-control paradigms (
1). Finally, at the trait level, mounting evidence suggests a U-shaped relationship, with individuals at both ends of the reward-sensitivity continuum showing inflammation. This pattern could reflect the tendency of individuals with high reward sensitivity to engage in inflammation-provoking behaviors—for example, consumption of foods rich in
trans fats and refined sugars. Together, these observations suggest that the neuroimmune network hypothesis should be elaborated to reflect the context dependence of reward-immune signaling.
The study has several limitations. First, its cross-sectional design precludes inferences about causality, a problem that could be partially addressed in multiwave longitudinal studies. Second, there is currently no technology to directly measure neural-immune crosstalk, so our inferences about its occurrence are necessarily indirect. The brain’s resident immune cells—microglia—appear to be central to this crosstalk (
5). As methods to image these cells’ functions develop, they will allow for direct evaluation of the framework’s key tenets. In the meantime, it would be valuable to more thoroughly explicate the neural basis of these results, using functional connectivity analyses to probe the broader cortico-amygdala and cortico-striatal circuits supporting threat and reward processing. It would also be valuable to replicate these findings with different paradigms, to determine whether they generalize to other threat and reward stimuli. Third, while consistent with the framework’s predictions, the interactions we observed were small to medium in terms of effect size, suggesting that to understand the health risks associated with poverty, a more thorough assessment of relevant exposures will be needed in future research. Of likely importance in this regard are the timing and duration of poverty, as well as presumptive mediators of its sequelae (e.g., stressors such as deprivation, violence, and discrimination). Finally, the study did not consider clinical outcomes, so it is unclear whether the hypothesized crosstalk accounts for health problems in the manner the framework suggests.
To summarize, these results provide initial support for the hypothesis that childhood stress amplifies crosstalk between peripheral inflammatory cells and brain regions involved in threat and reward. If substantiated, these patterns will have implications for understanding how early stressors, acting through neural-immune pathways, contribute to the development of a diverse set of health problems. With regard to mental health, this might take the form of a two-hit scenario, where stress-related inflammatory signaling increases risk for fear-related symptoms (e.g., vigilance, worry, rumination) by modulating amygdala circuity, and motivation-related symptoms by modulating striatal circuitry (e.g., substance misuse, anhedonia, mania). As the neuroimmune network hypothesis suggests, these neural and behavioral changes might, in turn, initiate a positive feedback loop that worsens psychiatric symptoms and extends them into other realms. Of course, longitudinal studies are needed to evaluate this scenario’s validity, but in the meantime it provides a framework for conceptualizing how vulnerability arises. In the long term, this work could facilitate a next generation of interventions that improve psychiatric outcomes by targeting brain-to-immune and/or immune-to-brain signaling.