Increasing evidence points to a role for the gut microbiome in modulating brain and behavior (
1). However, the underlying mechanisms by which microbes in the gut can influence central nervous system functioning are still unclear. The immune system is poised to be an important aspect in such signaling. Indeed, the gut is a strategic site for the training of the immune system, with gut bacteria educating immune populations and numbers (
2). Recently, it has been proposed that depression is associated with alterations in the stress response, inflammation, and gut microbiota composition (
3). The gut microbiome has thus become a crucial emerging piece in our understanding of the complex puzzle of depression. A growing number of studies show altered microbiome composition in patients with depression (
4) and that targeting the microbiome (psychobiotic approaches) may have beneficial effects (
5). Finally, preclinical studies have shown that the gut microbiome of individuals with depression can induce depressive-like behavior when transferred to animals (
4).
In recent decades, studies in both animals and humans have increasingly uncovered an intriguing involvement of inflammation in the etiology of depression. In fact, the levels of circulating proinflammatory factors, including T helper 17 (Th17) cells, have been reported to be altered in patients with depression (
6,
7). Conversely, blocking proinflammatory mediators may improve symptoms in patients who have higher baseline levels of inflammation (
8). However, the mechanisms underpinning how the microbiome and the immune system interplay in depression remains unknown. Quorum sensing is an important regulatory process for bacteria that enables cell-cell communication through the secretion of autoinducers to modulate and shape the cell density and gene expression in response to the environment (
9).
Writing in this issue of the
Journal, Medina-Rodriguez and colleagues (
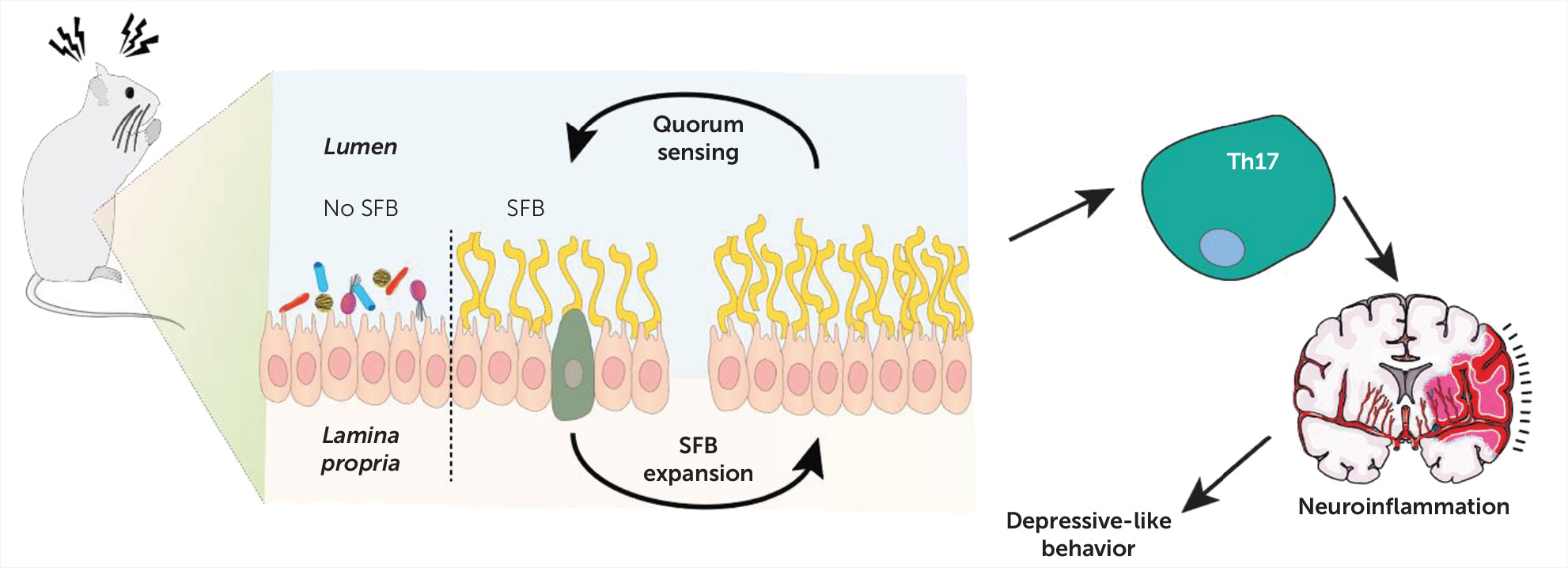
10) use an animal model to unveil a novel mechanism by which a quorum-sensing molecule—autoinducer-2 (AI-2)—that is produced by segmented filamentous bacteria (SFB) can increase susceptibility to depressive-like behaviors, in a Th17-dependent manner.
First, the authors investigate the relevance of different gut microbiome composition to the stress response and its associated depressive-like behavior. Genetically identical animals obtained from two different suppliers, the Jackson Laboratory (JAX) and Taconic Biosciences (TAC), that have long been known to harbor distinct gut microbiomes (
11) were exposed to a foot shock stress protocol, in order to induce learned helplessness. Interestingly, TAC mice were found to be more susceptible to learned helplessness than JAX mice, suggesting a potential involvement of the gut microbiome in mediating the development of this depressive-like behavior. Cohousing, a well-established manipulation for microbiome transplantation/exchange that takes advantage of grooming and coprophagic behaviors of mice, resulted in increased learned helplessness in JAX mice caged with TAC mice. This suggests that the transfer of gut microbiome from TAC mice aggravates sensitivity to learned helplessness or that, conversely, the gut microbiome in JAX mice confers resilience to stress. To pinpoint which bacteria could be responsible for this, the study authors retested TAC mice—which are more sensitive—to select a small fraction of mice that retained the learned helplessness phenotype after 3 to 9 weeks of retesting. Gut microbiome sequencing revealed that SFB levels were increased in mice that retained the phenotype after 4 weeks. This result thus supported the hypothesis that SFB could be able to enhance sensitivity to learned helplessness. Curiously, JAX mice, which are less susceptible to displaying learned helplessness, present extremely low values of SFB (
12). In fact, JAX mice cohoused with TAC mice display SFB levels closer to those of TAC mice. Next, to confirm the involvement of SFB in the development of learned helplessness, JAX mice were gavaged with SFB monocolonized feces, which resulted in sensitization to learned helplessness.
Together, these experiments established that the gut microbiome composition influenced sensitivity to foot shock–induced learned helplessness, suggesting that the presence of specific bacteria (SFB) increased susceptibility to learned helplessness. To tackle how these gut bacteria could modulate the depressive-like behavior response, the authors next set out to explore quorum sensing in the context of this depression model. Communication via quorum sensing involves the secretion of autoinducer molecules that shape bacterial population numbers and gene expression (
9). The main quorum sensing molecules—AI-2, sc-AHL, and lc-AHL—were thus analyzed in the feces of JAX, TAC, and cohoused animals. The results clearly showed that AI-2 is increased in TAC mice and in JAX mice cohoused with TAC mice and that AI-2 may be preferentially expressed in SFB. Furthermore, TAC mice showed increased levels of AI-2 upon foot shock stress exposure. In line with these results, administration of AI-2 to mice altered immobility time in the tail suspension test, while interfering with sociability.
SFB is known to stimulate the generation of Th17 cells (
12), which have been reported to bolster depressive-like behaviors (
13,
14), and Th17 cells have been shown to concentrate in the hippocampus of mice that develop learned helplessness (
15). Remarkably, the transfer of CD4+ cells that present specialized T cell receptors for SFB led to learned helplessness and also to the accumulation of Th17 cells in the hippocampus of recipient mice. This means that SFB can modulate Th17 cells in the context of a mouse model of depression.
To understand whether SFB enhances learned helplessness and hippocampal Th17 accumulation through AI-2, the authors then administered the AI-2 molecule, which resulted in increased accumulation of Th17 in the hippocampus. Next, the authors took advantage of the RORc(γT)
+/GFP mouse model, which has dysfunctional Th17 cells and has previously been shown to be resistant to the development of learned helplessness but becomes susceptible upon adoptive transfer of Th17 cells (
13). In the present study, Medina-Rodriguez and colleagues demonstrated that the depressive-like phenotype observed upon treatment with AI-2 was abrogated in RORc(γT)
+/GFP mice, establishing that Th17 cells are necessary to modulate AI-2-evoked depressive-like behavior.
Th17 cells are largely located in the small intestine, and SFB has been shown to enhance Th17 differentiation at this site through the SAA1-2/IL-22 pathway (
12). AI-2 administration and learned helplessness after 4 weeks of stress resulted in increased intestinal levels of SAA1 and SAA2, but not SAA3 and IL-22. Moreover, RORc(γT)
+/GFP mice that are resistant to learned helplessness display lower levels of SAA1 and SAA2, and injection of SAA2 is sufficient to sensitize these mice to learned helplessness. Overall, this set of experiments details a network through which SAA1 and SAA2 affect Th17 cells, which affects depressive-like behavior.
The final part of the story is quite intriguing. Oleic acid is a natural compound that has been found to interfere with AI-2 activity (
16). Here, the authors showed that it diminished susceptibility to learned helplessness in TAC mice while modulating SAA1 and SAA2 levels in the gut. Additionally, oleic acid treatment reduced accumulation of Th17 cells in the hippocampus, suggesting that perhaps dietary interventions can effectively modulate this pathway and hence ameliorate depressive-like behavior.
Although all of this research is in animal models, the authors sought to investigate whether there was any translational relevance. Thus, in a small sample of patients with depression, IL-17-A fecal levels were found to be increased, as well as SFB abundance and SAA levels, which parallels some of the findings in animals and suggests the involvement of the described pathway in a clinical setting. Future validation of this finding in larger cohort and in a longitudinal manner is needed.
Inflammation and the gut microbiome are key players that can no longer be overlooked in the field of neuropsychiatry. That the periphery provides important inputs in the central nervous system is now unquestionable; however, the underlying mechanisms that explain the functioning of this complex machinery remain highly underexplored. The understanding of these pathways will promote more targeted approaches that will improve therapeutic strategies and ultimately benefit patients. The study by Medina-Rodriguez and colleagues elegantly characterizes a mechanism (see
Figure 1) through which the miscommunication between the gut microbiome and the immune system caused by stress perniciously affects the hippocampus, an essential limbic structure, which ultimately unfolds as depressive-like behavior. These observations will undoubtably contribute to the advance of the field, providing a mechanistic pathway worthy of further exploration.
Acknowledgments
The authors thank Dr. Kenneth O’Riordan for assistance with Figure 1. Prof. Cryan is funded by Science Foundation Ireland SFI/12/RC/ 2273_P2, the Saks Kavanaugh Foundation, EU H2020 project DLV-848228 DISCOvERIE, and Swiss National Science Foundation project CRSII5_186346/NMS2068.