Major depressive disorder (MDD) is a common psychiatric syndrome with considerable disease burden. It is a leading contributor to disability, suicidality, and productivity loss in the global economy (
1,
2). Individuals with MDD exhibit varying symptom profiles (
3), which are sometimes opposing—for example, some show symptoms of insomnia and weight loss while others experience hypersomnia or weight gain. Standard typology partly captures this heterogeneity; however, it remains elusive whether it adequately captures etiological heterogeneity. Given the relatively low heritability of 30%–40% (
4), it is imperative to investigate the genetic characterization of MDD heterogeneity and to identify more homogeneous patient subgroups.
Recent studies using large-scale samples with standardized phenotyping and genotyping have contributed to a better understanding of genetic heterogeneity in MDD (
5,
6). For example, in our previous work, we used UK Biobank data to systematically compare the genetic architecture of 16 MDD subtypes (
6). We showed that clinical subtypes were genetically more homogeneous than all MDD together. However, that study relied mostly on self-report data and retrospective recall of depressive symptoms. Also, given the known “healthy volunteer” bias in the UK Biobank (
7), a key limitation was that individuals with more severe illness were underrepresented. Earlier family-based studies on genetic contributions to MDD subtypes were limited by small sample size, few subgroups, and inconsistent phenotype definition mainly based on self-report data. Findings on genetic heterogeneity in MDD remain inconclusive and debatable (
4,
8,
9). Therefore, to untangle the genetic heterogeneity of MDD, more evidence is needed, particularly for the patient subgroups with the greatest clinical severity and the highest disease burden.
Using register data from the entire Swedish population, we sought to strengthen the evidence on the genetic heterogeneity of MDD. Among individuals with specialist-diagnosed MDD, we studied 18 subgroups based on nine comparison groups defined by clinical and psychosocial features. Using the genealogical information available in the Swedish registers, we applied a sibling-based design and used classic quantitative genetic models to investigate the genetic components of these subgroups. To clearly demonstrate the synthesized evidence, we compared the resultant pedigree-based estimates with the estimates based on single-nucleotide polymorphisms (SNPs) from our previous UK Biobank study.
Results
From 1,500,713 individuals born in Sweden between 1977 and 1993, we randomly selected one pair of full or maternal half siblings from each family, with the siblings born ≤10 years apart. The final sample included 838,990 individuals who formed 419,495 unique sibling pairs (395,531 full sibling pairs; 23,964 maternal half sibling pairs) (see Figure S1 in the
online supplement). Among these, we identified 46,255 individuals with MDD (5.5%). Age at first MDD diagnosis ranged between 7.8 and 36.9 years (mean=23.2 years). Compared with those without MDD, the MDD group had a higher proportion of females (MDD group: 63.0%; non-MDD group: 47.8%). The two groups did not differ in mean age at end of follow-up (MDD group: mean=27.7 years, SE=4.3; non-MDD group: mean=28.0 years, SE=4.2). The characteristics of each subgroup are summarized in Table S5 in the
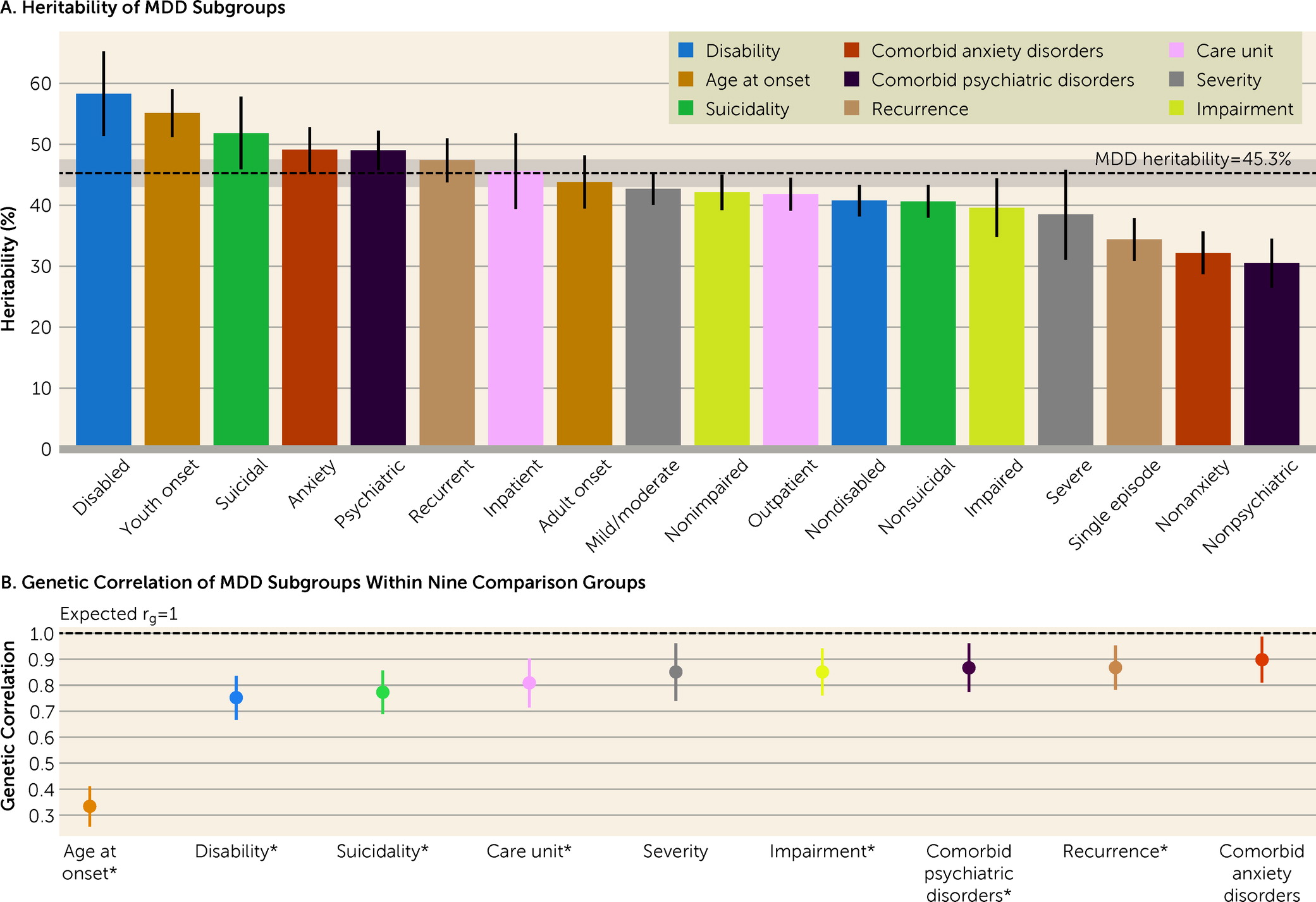
online supplement. The heritability estimated for MDD was 45.3% (95% CI=43.0–47.5), similar to previous estimates based on the same population (
17).
The number of individuals in the 18 MDD subgroups ranged between 6,857 (14.8% of the MDD sample) and 39,398 (85.2%) (
Table 1). For the subgroups, the heritability estimates ranged from 30.5% to 58.3% (
Figure 1A; see also Table S6 in the
online supplement). The disabled and youth-onset subgroups showed significantly higher h
2 than the overall MDD sample, while the subgroups with single-episode MDD and without comorbid anxiety disorder or other psychiatric disorders showed significantly lower estimates (see Table S7 in the
online supplement). The r
g estimates within comparison groups ranged from 0.33 to 0.90 (
Figure 1B). Seven of nine r
g values were statistically significantly different from 1 (p<5.56×10
−3) (see Table S8 in the
online supplement).
Among all the subgroups studied, the subgroup with disability (based on early retirement) had the highest h
2, at 58.3% (95% CI=51.4–65.2). This estimate was significantly higher than that for the nondisabled subgroup (h
2=40.8%, 95% CI=38.20–43.3; p=2.63×10
−6 for difference in estimates) (see Table S7 in the
online supplement). The r
g between these two subgroups was significantly lower than 1 (r
g=0.75, 95% CI=0.67–0.84) (
Figure 1B; see also Table S8 in the
online supplement). For the disabled subgroup, there was suggestive evidence for the influence of a C component (22.3%, 95% CI=2.3–42.3, p=0.03) (see Table S6 in the
online supplement).
For the comparison groups based on age at first diagnosis, youth-onset MDD was more heritable than adult-onset, with h
2 estimates of 55.1% (95% CI=51.2–59.0) and 43.8% (95% CI=39.5–48.2), respectively. The r
g between the two subgroups (r
g=0.33, 95% CI=0.26–0.41) was the lowest among the nine comparison groups (
Figure 1; see also Tables S6–S8 in the
online supplement).
The suicidal subgroup showed a significantly higher h2 than the nonsuicidal subgroup (51.8% vs. 40.6%, p=8.05×10−4), and their rg was 0.77 (95% CI=0.69–0.86).
The subgroups with comorbid anxiety disorder and with other psychiatric disorders showed higher h
2 estimates than the subgroups without these comorbidities (
Figure 1A; see also Table S7 in the
online supplement). However, the r
g values within these two comparison groups were high, at 0.90 (95% CI=0.81–0.99) between the subgroups with or without comorbid anxiety disorder, and 0.87 (95% CI=0.77–0.96) for the ones with or without other psychiatric disorders.
Similarly, recurrent MDD was more heritable than single-episode MDD (47.4% and 34.4%, respectively; p=3.84×10
−7). The r
g of these two subgroups was 0.87 (95% CI=0.78–0.95) (
Figure 1).
The inpatient subgroup (i.e., individuals who had been hospitalized for MDD) showed a slightly higher h
2 than the subgroup treated only in outpatient care (45.6% [95% CI=39.4–51.8] compared with 41.8% [95% CI=39.1–44.5]), but the difference was not statistically significant. The r
g within this comparison was significantly lower than 1 (r
g=0.81, 95% CI=0.72–0.90) (
Figure 1; see also Tables S7 and S8 in the
online supplement). By including only a primary diagnosis of MDD in the inpatient subgroup, we may have missed relevant MDD cases in which the patient was hospitalized with suicide as the main diagnosis. We conducted sensitivity analyses where 287 MDD cases with a primary diagnosis of suicide were added to the inpatient subgroup. The results were consistent with the primary results (see Table S9 in the
online supplement).
The two comparison groups based on severity and impairment showed a high within-comparison r
g of 0.85 (
Figure 1B). The h
2 estimates for the severe and impaired subgroups were slightly lower compared with those for their counterparts, albeit with largely overlapping confidence intervals (
Figure 1A; see also Table S7 in the
online supplement).
Considerable sample overlap (mean overlap, 48.3%; range, 6.4–93.6; see Figure S2 in the online supplement) and phenotypic correlations (range, −0.55 to 0.55; see Figure S3 in the online supplement) contributed to the high rg estimates between subgroups across comparison groups (mean=0.89; see Figures S3 and S4 in the online supplement). Nonetheless, 48% (69/144) of the rg values had confidence intervals that did not include 1. Some comparison groups showed different patterns of cross-comparison rg. For example, as expected, the inpatient subgroup had a strong genetic correlation with the severe MDD subgroup (rg=0.91), while the outpatient subgroup had a strong genetic correlation with the mild/moderate MDD subgroup (rg=0.99). Compared with the adult-onset subgroup, the youth-onset subgroup had a stronger genetic correlation with all but the impaired subgroup (rg>0.7) (see Figure S4 in the online supplement).
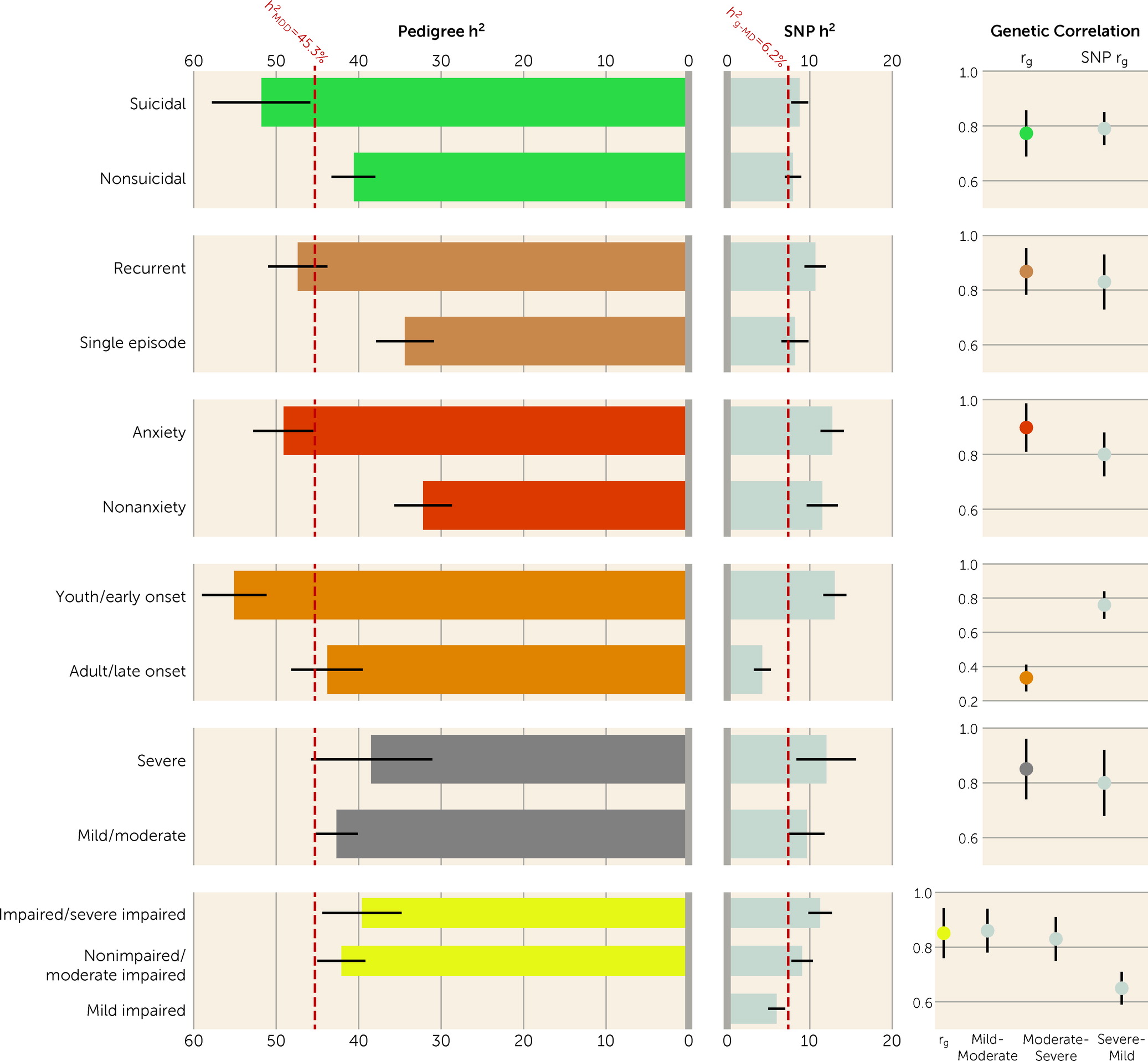
Heterogeneity of MDD Subgroups Observed Using Pedigree and Genomic Data
We compared the h
2 and within-comparison-group r
g values estimated using pedigree data (this study) with the estimates from genomic data (our previous study, using the UK Biobank cohort) (
Figure 2). Among the six overlapping subgroups, SNP h
2 was about one-tenth to one-third of h
2 across subgroups. In general, patterns of heritability estimates were similar between the two studies (
Figure 2; see also Table S10 in the
online supplement). In both studies, the early/youth-onset subgroup was significantly more heritable than the late/adult-onset subgroup (
Figure 2; see also Table S6 in the supplement of reference
6). Using pedigree data, we observed significant differences in h
2 based on more clinical indices, including suicidality, recurrence, and comorbid anxiety disorder, for which we did not find the same results in the UK Biobank data. Contrary to the h
2 estimates, the SNP h
2 values for the severe and impaired subgroups were higher than their counterparts in the UK Biobank data.
The rg estimates were similar between the two studies, with mean values of 0.76 and 0.79 for the pedigree and genomic data, respectively. In both studies, rg was significantly lower than 1 for subgroups within the comparisons of age at diagnosis/onset, suicidality, recurrence, and impairment. However, the rg between youth-onset and adult-onset MDD from the present study was significantly different from that of early-onset and late-onset MDD from the UK Biobank study (0.33 and 0.76, respectively; p=3.97×10−13).
Discussion
In this study, we used Swedish national registers to investigate and contrast genetic components of 18 MDD subgroups. The range of heritability of these MDD subgroups was 30.5%–58.3%, nearly a twofold difference. The majority of genetic correlations within comparison groups (range, 0.33–0.90) significantly deviated from 1, suggesting differences in their underlying genetic risk factors. Furthermore, we followed up on our previous work and compared these pedigree-based estimates with the SNP-based estimates from the UK Biobank. In both studies, we found that, in general, subgroups with more severe manifestation were more heritable than their counterparts, and genetic correlations from the two studies were comparable (mean correlations of 0.76 and 0.79 for pedigree- and SNP-based, respectively). These findings strengthen the current evidence that subgroups of MDD differ in their genetic components.
It has been challenging to characterize genetic contribution to MDD heterogeneity. The large samples from Sweden and the UK Biobank provide unique opportunities to systematically compare genetic components of MDD subgroups. The present study, however, differs from our previous UK Biobank study in several important respects. First, here we used a Swedish population-based cohort, as compared to a more selected population in the UK Biobank (
7). With specialist-treated MDD extracted from patient registers, the samples used here represent a patient population with more severe illness and greater functional impairment, thereby addressing a key limitation of the previous study. Second, in the previous study, which was primarily based on retrospective self-reported symptoms, we focused on symptom-based subtypes, whereas in this study we focused on the subgroups with differential psychosocial function and disease burden. Third, the previous findings based on genomic data were reexamined here using classic quantitative genetic modeling. Across the six overlapping subgroups, the SNP-based heritability accounted for only a small percentage (10%–30%) of pedigree heritability. The difference is likely due to two main factors. First, SNP-based heritability only captures the genetic contribution explained by common genetic variants that are tagged on genotyping arrays. Second, the dissimilarities between the two studies, including phenotype definitions and sample ascertainment (i.e., specialist-treated MDD versus self-reported major depression) may also contribute to the variation. Notwithstanding the differences, the findings from the two studies were largely consistent; together they provide the most comprehensive overview of genetic heterogeneity in MDD to date. Identifying more heritable subgroups would increase GWAS power. The present study found up to 1.5 times higher heritability in subgroups than in all MDD; assuming the SNP heritability of 9% for all MDD (
24) and a similar fold increase in subgroups’ SNP heritability, this would reduce sample size by approximately one-third while maintaining power to detect significant loci (
25).
Notably, this study extends evidence for subgroups with greater functional disability and disease burden. We used medical and social benefit records to examine clinical and psychosocial features as a means of defining MDD subgroups. We found that the disabled subgroup, as indexed by early retirement, was the most heritable subgroup among all those studied. Compared with their counterparts, heritability was significantly higher for the MDD subgroups with disability, youth onset, suicide attempt or death by suicide, comorbidity with anxiety disorder or another psychiatric disorder, and recurrence. These results were largely consistent with previous analyses using polygenic risk scores, which showed that subgroups with youth onset, recurrence, and comorbid anxiety disorder had a higher genetic burden of common risk alleles for MDD than the later-onset, single-episode, and nonanxiety MDD subgroups, respectively (
5,
26). On the other hand, severity and impairment within MDD in this study appeared to be less useful differentiators. This might be due to the limited variability in these two indicators in specialist-treated MDD. Nevertheless, these results underscore the importance of studying the patient subgroups on the far end of the spectrum of functional disability, especially using genetic approaches.
Despite the evidence supporting genetic heterogeneity in MDD, the relatively high genetic correlations clearly demonstrated that the genetic components of subgroups are only partially distinct. Except for the age-at-diagnosis comparison, all pairwise genetic correlations were between 0.75 and 0.90. The subgroups of youth-onset MDD (defined as age ≤21 at the first specialist MDD diagnosis) and adult-onset (age ≥25) showed a much lower genetic correlation of 0.33. This estimate was also substantially lower than our UK Biobank estimate of 0.76 between early-onset depression (defined as age at first experience of a ≥2-week episode of cardinal symptoms at age ≤30 years) and late-onset depression (≥44 years). It should be noted that the two studies had major differences in phenotype definitions (age at diagnosis vs. age at onset) and cutoffs (youth- or adult-onset vs. early- or late-onset), especially given the fact that many individuals with MDD do not seek treatment until well after symptom onset (
27). These differences are likely to have led to discrepancies in the results. Both our own work based on the UK Biobank (
27) and the Australian Genetics Study of Depression (
5) showed that the polygenic risk scores of MDD were the highest among individuals with an early onset age and steadily declined until an onset age of around 30. Youth depression has been associated with subsequent somatic diseases and premature mortality (
28). Thus, it might be meaningful and clinically important to identify specific genetic risk factors underlying this subgroup.
Interpretation of the study results is subject to several limitations. Changes in clinical practice during the study follow-up period constitute a possible limitation of using register data. Changes in practice may have led to differences in MDD diagnoses in individuals across different years. To limit potential bias owing to this factor, we used a sibling design (as opposed to familial relations across generations) with sibling pairs born ≤10 years apart and adjusted for the linear and quadratic effects of birth year in all models except those for the subgroups based on age at diagnosis. We were not able to appropriately account for potential diagnostic changes over calendar time in the comparison of youth-onset versus adult-onset MDD. Second, although national register data allowed us to study subgroups in large samples, we still lacked sufficient sample sizes to study rare subtypes, such as psychotic MDD (less than 4% of all MDD cases in the Swedish register), and lacked fine-grained clinical information on symptoms and treatment response to investigate important subtypes such as atypical or treatment-resistant MDD. These represent areas of interest for future studies. Third, we do not have information to identify the specific indication for receiving sickness benefits and early retirement pensions. Fourth, we may have missed MDD cases or misclassified subgroups beyond the end of follow-up. However, our data covered the peak onset age of MDD for the investigated cohort, and for the majority of MDD cases, we have sufficient follow-up time to correctly assign subgroups (median=5.1 years since the first MDD diagnosis, IQR=2.5–7.8 years). Moreover, the results from this study were similar to our previous findings in the UK Biobank, regardless of the differences in birth cohort and follow-up. Finally, despite many advantages of register data, we cannot rule out the potential impact of the differences in the phenotypic reliability between subgroups on the observed findings.
For all subgroups, we did not find any statistically significant contribution of the common environment (C) to the liability of the disorders. Our simulation (see the Supplementary Results section in the online supplement) showed that with the sample size used here, we had sufficient power (>80%) to detect a significant C component of ∼10% and above. However, the power might be limited if C is of a smaller magnitude (e.g., we had less than 60% power to detect a C component of <8%) (see the Supplementary Results section in the online supplement). Our models relied on the assumption of no gene-environment interplay, which, if violated, could affect the estimates.
Treating MDD as a single form limits the potential for personalized treatment. Progress will depend on patient stratification based on clinical characteristics, or on the omics features promised by advanced technologies. This study has produced important insights into the genetic heterogeneity of MDD and a deeper etiological understanding of MDD clinical subgroups. These insights will encourage future studies to identify specific genetic factors and biomarkers, and to map subgroups to outcomes, thereby accelerating research on novel and optimized treatment tailored to specific patient groups.