The Genomics of Bipolar Disorder
A child of an affected parent has about a tenfold increased risk of developing BD, and twin studies estimate a heritability of 0.7–0.8 (
1). There is no evidence for Mendelian inheritance or for genes of major effect. Instead, as with most psychiatric disorders, there are multiple susceptibility loci, each of small effect, which genome-wide association studies (GWAS) are beginning to identify. Several GWAS, and meta-analyses thereof, have been carried out since 2007;
Table 1 lists the loci and implicated genes that have emerged to date. The combined sample sizes remain small by GWAS standards, and more loci remain to be identified; indeed, the forthcoming Psychiatric Genomics Consortium analysis, comprising over 20 000 BD cases and 30 000 controls, identifies 19 significant loci, including 12 novel ones. Initial exome and genome sequencing data suggest that rare deleterious variants also have a role in some BD cases, but their identity and overall contribution to the disorder remain unclear (
12–
14). Within BD, there is modest clinicogenetic heterogeneity, for example, based on the predominant symptoms, or between
bipolar I and
bipolar II subtypes (
15). However, there is little evidence for BD-specific genes; joint GWAS analyses show substantial commonalities in risk loci for BD and schizophrenia (
16), as well as significant overlap with other major psychiatric disorders (
17) and with intermediate phenotypes, including circadian traits (
18,
19). One distinction between schizophrenia and BD is that copy number variation is much less prominent in the latter (
20).
Although the genomics of BD are in their infancy, efforts have begun to understand the biological basis for the associations identified to date. Interest has centred on two genes (
CACNA1C and
ANK3) because of what was already known of their functions.
CACNA1C is discussed in detail below.
ANK3 encodes ankyrin G, which couples axonal voltage-gated sodium channels to the cytoskeleton and also has roles in dendrites and glia; another risk gene,
TRANK1, contains multiple ankyrin repeat domains, suggesting some shared functions. Complementing the focus on specific risk genes, the first attempts have been made to identify the pathways that they influence. Using data from four of the GWAS, Nurnberger
et al. (
21) reported six pathways that showed replicable association with BD, involving glutamate and calcium signalling, second messengers, and hormones. Together, these findings support the possibility that BD is, at least in part, an ion channelopathy (
22), in which aberrant calcium signalling is important (
23).
Calcium Signalling in Bipolar Disorder: Linking Genetics, Pathophysiology, and Therapeutics
Calcium dysregulation has long been implicated in BD, based primarily on
ex vivo studies in cells taken from patients and controls. The findings are disparate, but on balance indicate that measures of intracellular calcium signalling are increased in BD, especially after stimulation (reviewed in (
23,
24)). The abnormalities appear largely independent of current mood state (i.e., they are trait rather than state related). Moreover, they are attenuated by lithium, which is used in the treatment of the disorder (
Box 1). Despite the many uncertainties, these findings led to L-type voltage-gated calcium channel (VGCC) antagonists, with existing indications in angina and hypertension, being evaluated for the treatment of BD (
25). Antiepileptic drugs, such as pregabalin, which act via VGCC α
2δ subunits (
Box 2) have also been tested (
26), and lamotrigine, another antiepileptic drug that may block calcium channels, among its various actions (
27), is an effective treatment for bipolar depression (
28).
The results of the recent genomic studies strongly suggest that the involvement of calcium signalling in BD is at least partly causal (
29), and have rekindled attempts to explain more precisely the nature of the alterations, not least because this may provide clues to more-effective and tolerable drug strategies to normalise them (
30). However, the discovery of genetic variants is only the first step, and provides many more questions than answers. Calcium signalling offers an informative exemplar to highlight the opportunities and complexities associated with moving from psychiatric genomic discoveries to pathophysiological insights and therapeutic advances (
31,
32).
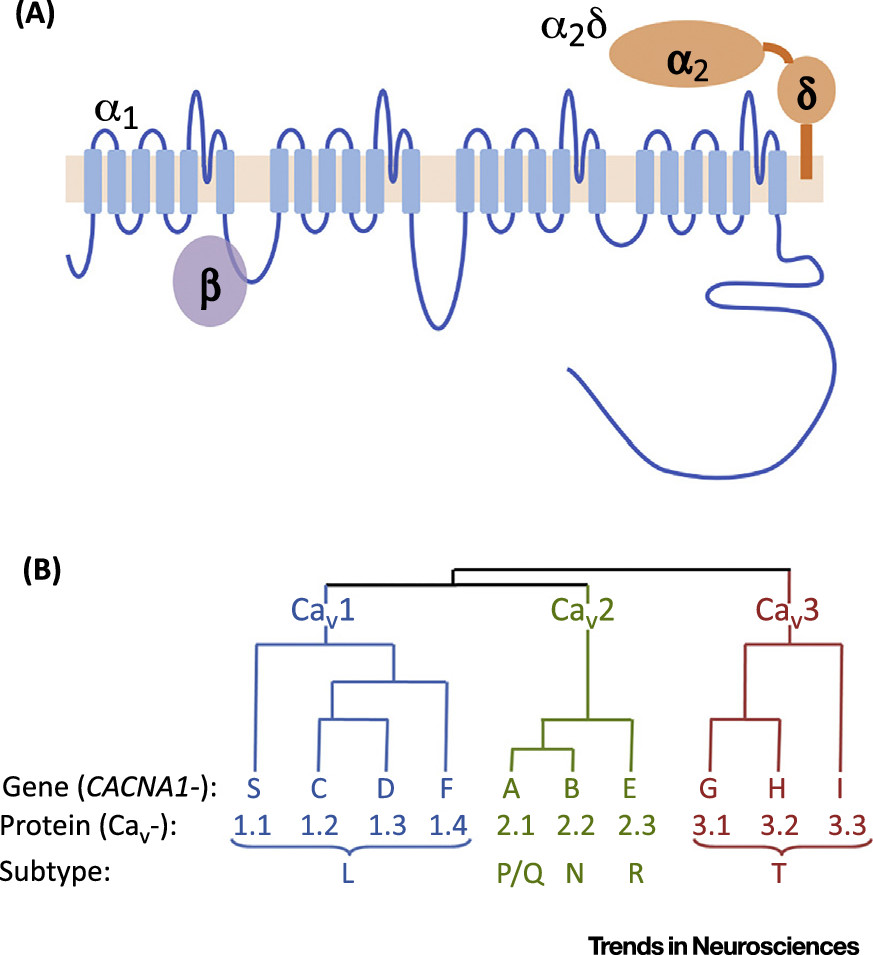
Genomic data provide a starting point to identify the molecules involved in the core ‘calcium pathophysiology’ of BD. They focus attention on the VGCCs, especially of the L-type, and their accessory subunits (encoded by the
CACNx genes;
Box 2). As indicated above, the best evidence is for
CACNA1C (encoding the α1 subunit of Ca
v 1.2), but pathway analysis also suggests a role for
CACNA1D and
CACNB3 (
29,
33), and other VGCC genes are implicated in BD by rare variant studies (
13). Of note, apart from BD, CACNA1C is associated with schizophrenia (
34) and major depression (
35), and CACNB2 confers susceptibility to multiple psychiatric disorders (
17). Involvement of VGCC genes has also been reported in large-scale genomic studies of BD-relevant phenotypes, such as working memory performance and the associated patterns of brain activation (
36), as well as in general cognitive functioning (
37). There are also smaller candidate gene studies that suggest effects of CACNA1C genetic variation on brain imaging phenotypes (
38,
39) and on cognitive domains, such as reward responsiveness (
40).
Identification of the molecular basis for disease associations is a key step in understanding the mechanisms linking VGCC genes with BD. The VGCC loci revealed by GWAS are noncoding, and while large-scale exome studies may identify rare variants that disrupt the coding sequence of VGCCs, it is unlikely that the BD-associated GWAS loci tag as-yet-unidentified, disease-causing mutations. Instead, they probably act by influencing aspects of gene expression, including methylation (
41), alternative promoter usage, and RNA splicing (
42). In the case of
CACNA1C, the index risk polymorphism for BD (rs1006737) is located in the third intron. On balance, the available data indicate that the risk allele is associated with enhanced expression and activity of the gene product (
43,
44), but there are conflicting findings (
45,
46), precluding firm conclusions. Some of the variability in these results may be due to differences in the effect of rs1006737 on
CACNA1C expression between brain regions. Inconsistencies may also result from the risk single nucleotide polymorphism (SNP) differentially altering the abundance of particular splice variants, as has been observed for other BD-relevant genes, including
ANK3 (
47) and
ZNF804A (
48). In the case of
ANK3, there is evidence that the shift in isoform ratios has functional consequences for neuronal physiology (
49). Efforts to identify and understand whether altered splicing is also relevant for
CACNA1C and other VGCCs are hampered by the limited information about their isoform profile in the human brain (
Box 2). This information is critical since splicing patterns are poorly conserved and the brain shows one of the greatest diversities of alternative splicing (
50), meaning that the current data, which pertain to other species and tissues, are insufficient.
While the identification of specific VGCC subunits and isoforms that mediate BD risk is important, better understanding of the underlying biology is also crucial. This requires the use of appropriate cellular and animal model systems, as well as
in vivo approaches in humans. For cellular analyses, in addition to standard cell lines, which are useful for studying the function of individual genes, induced pluripotent stem cells (iPSCs) may prove valuable. Indeed, iPSC-derived neurons have already provided intriguing data to support the presence of cellular BD-related phenotypes, including alterations in calcium signalling (
Box 3).
The molecular findings can also help guide the development of rodent models overexpressing or lacking specific VGCC genes (
51–
53) or splice variants thereof (
54), in which their functional impact can be studied in the intact animal. For example, embryonic deletion of Cacna1c from forebrain glutamatergic neurons in mice produced BD-relevant behavioural and cognitive effects and an increased susceptibility to stress, whereas the same deletion during adulthood caused a lesser and, in some instances, opposite phenotype (
51). This effect of developmental stage on the phenotype is intriguing, given the characteristic early adulthood age of BD onset, and its childhood antecedents (
Box 1). In a separate study, the phenotype of Cacna1c-deficient mice could be rescued using a small-molecule inhibitor of the translation initiation factor eIF2α (
52), giving clues as to the possible intervening biochemical mechanisms.
In parallel with these various genetically driven approaches, pharmacological investigations of VGCCs in humans are possible because of the existing L-type VGCC antagonists, which can be used as experimental tools and for proof-of-principle studies. Their availability is a distinct advantage compared with most other genetically supported therapeutic targets in BD, for which no such drugs are available. To date, the clinical trial data linking L-type VGCC blockade with therapeutic outcome in BD (and, indeed, their psychiatric effects more generally) are wholly inconclusive (
25). Hence, a priority is to investigate in detail the impact of brain-penetrant VGCC blockers on BD-relevant phenotypes, including detailed measures of mood, cognition, sleep, and brain activity (
55), and with the incorporation of genotype as a factor (
56). The potential psychiatric effects of VGCC antagonists can also be assessed using routinely collected clinical data; for example, a study of electronic medical records in Scotland reported higher hospital admission rates for depression in patients given VGCCs compared with those prescribed other antihypertensives (
57).
In summary, a range of methods are needed to make the most of the genomic discoveries in BD, to understand their molecular mechanisms and implications for cellular and systems functioning, and to evaluate their therapeutic potential. The conceptual approach is illustrated in
Figure 1 (Key Figure), which uses VGCCs as the exemplar.
Persistent Mood Instability
Notwithstanding the many challenges, the longitudinal, long-term collection of data using digital approaches has already contributed to a renewed focus on the ‘real-world’ phenotype of BD. One example is the appreciation that many patients have chronic mood instability (also called
affective lability), persisting during
euthymia. This contrasts with the simplistic textbook description of BD as comprising periods of normal, stable mood in between the episodes of depression and mania (
Box 1). Although this reality was already appreciated by experienced clinicians (
69,
70), it is the use of digital methods that has led to a greater awareness of mood instability, which in turn has encouraged research to understand its origins and significance. Here, we briefly review some of these implications.
Mood (in)stability is a continuous variable, and is present to varying extents in healthy individuals (
71,
72). Therefore, it is a useful phenotype for studies seeking to identify mechanisms underlying mood (dys)regulation
per se, and a range of experimental approaches are being taken. For example, it can be viewed from a computational perspective, with models showing how mood instability interacts with reward sensitivity to alter performance (
73). One can also ask how mood instability relates to variation in neural activity across a range of temporal resolutions, using functional MRI (
74) and magnetoencephalography (
75). In addition, mathematical analyses, such as those outlined in
Box 4, can help explain the relationships between fluctuations in mood and other parameters, ranging from environmental exposures to behavioural variables (
66,
67).
Mood instability is prominent not only in BD, but also in several other psychiatric disorders, such as attention deficit disorder, borderline personality disorder, and schizophrenia (
76,
77). It is therefore a trans-diagnostic construct, compatible with the NIMH Research Domain Criteria project (
78). Thus, understanding the origins and mechanisms of mood instability, and the biological and behavioural sequelae of interventions which modify it – is therefore likely to have value beyond BD. Indeed, mood instability is a moderately heritable trait in the general population, and the first genome-wide significant loci have been reported (
79). Apart from studying the implicated molecules and pathways in their own right, it will be of interest to investigate the extent to which the risk loci for mood instability overlap with those for BD specifically, and other disorders involving mood instability. Moreover, the specific characteristics of mood instability (e.g., its frequency, amplitude, or impact on behaviour), while partially overlapping, may differ sufficiently between disorders (
80) to help gain traction on the underlying neural mechanisms, providing further clues to the biological bases of these illnesses and leading to refinements in classification.
Finally, mood instability has potential immediate value as a therapeutic target. First, it may provide a way to screen novel treatments for BD more rapidly and cost-effectively than conventional clinical trials, in which treatment or prevention of depressive or manic episodes are the usual outcomes, requiring prolonged periods of observation. In this sense, early mood stabilisation could prove to predict therapeutic efficacy in the way that rapid effects on emotional processing predict subsequent therapeutic response in unipolar depression (
81). Moreover, the rapid effects of antidepressants on emotional processing are seen in euthymic subjects as well as in those with depression; similarly, new treatments for BD, such as novel VGCC-acting drugs, could be screened in patients who have unstable mood but who do not have a formal diagnosis. Second, stabilisation of mood may be beneficial in its own right, above and beyond simply preventing clinical episodes of depression and mania, because mood instability independently predicts poor functional recovery in BD, and worse outcomes of various kinds (
76,
82–
84). In addition, since mood instability often occurs before the onset of BD (
85,
86), such treatments might have a preventative role.
In summary, the emergence of persistent mood instability as a construct illustrates how digital methods can highlight neglected or hard-to characterise psychiatric phenomena, and stimulate a range of studies into their origins, mechanisms, and therapeutic implications. Comparable approaches are being taken to other components of BD, such as circadian dysregulation and reward sensitivity. Although the work is at an early stage, it is likely ultimately to alter the view of the core phenotype of BD, with a greater role for biological and quantitative data in BD diagnosis, prognosis, and management.
Concluding Remarks
Our understanding of BD remains frustratingly limited. It continues to be a descriptive syndrome, since we lack sufficient knowledge to allow its characterisation or conceptualisation based on aetiology or mechanism. Certainly, many questions remain (see Outstanding Questions). However, there are reasons for optimism. First, the discovery of some of the BD risk genes has the potential to revolutionise our understanding of its pathogenesis and neurobiology. Second, the use of digital technologies and remote sensors, coupled with advanced analyses of the resulting data, is already allowing a more-quantitative, longitudinal approach to the BD phenotype. This raises the potential for better prediction of an individual’s clinical course, and also provides a more-sophisticated phenotype for behavioural and biological studies. In both of these domains, a common feature is the increasing importance of ‘big data’, whether in terms of the genomic studies, or the multidimensional data streams captured by digital devices. Third, although not discussed here, structural and functional brain imaging is helping to identify the key neural circuits of BD and may also have diagnostic and prognostic value (
2–
4,
87–
90).
The ultimate goal of these contemporary approaches is to allow a more scientifically informed, evidence-based approach to how BD is classified, measured, and treated (
91). Possible changes include redrawing or removing the diagnostic boundaries between BD and other disorders involving lability of mood, emotion, and behaviour; the inclusion of behavioural and physiological correlates of mood and mood instability (or other symptoms) into clinical practice; a focus on identifying interventions that can stabilise mood independent of the underlying diagnosis; and new, genetically informed treatments, such as those targeting VGCCs.
It is not always appreciated that BD causes a global health burden comparable with that of schizophrenia (
92), yet it has attracted considerably less research funding and interest from policy makers (
93). We would argue that, for various reasons, BD currently has a greater potential for transformative advances. More generally, BD is a useful case study for illustrating how psychiatric disorders are belatedly embarking on the journey from being descriptive syndromes towards more neurobiologically grounded, quantitative, and digital phenotypes (
94). Despite the many difficulties this process entails, it is not unreasonable to hope that it will be successful, and accompanied by the development of more rational, effective, and personalised treatments.