Telomeres are repetitive hexa-nucleotide sequences that form the ends of eukaryotic chromosomes.
1 Telomeres shorten a few base pairs at every cell cycle, due to the end replication problem.
2 Studies have shown that reduced telomere length in peripheral blood is associated with illnesses of aging phenotypes, such as dementia,
3 and DNA repair deficit disorders, such as ataxia telangiectasia (AT).
4AT is an autosomal recessive disorder characterized by neurological deterioration, immunodeficiency, and premature ageing. AT mutated loss also leads to intrinsic mitochondrial abnormalities in thymocytes, such as elevated reactive oxygen species, increased aberrant mitochondria, high cellular respiratory capacity, and decreased mitophagy.
5,6 Although the telomerase activity is well preserved in the blood cells, shortened telomeres have been observed in lymphocytes in patients with AT.
4 A faulty telomere repair process or a separate accelerated telomere shortening process has been hypothesized to be responsible for these shortened telomeres.
Huntington’s disease (HD) is an autosomal dominant neurodegenerative disorder with a progressive, selective neural cell loss, and atrophy in the caudate and putamen, caused by a glutamine repeat expansion in the huntingtin protein. A number of cellular processes are disrupted by this change in huntingtin, and its pathological effects are still being investigated. Using striatal neuronal cell lines from wild-type and HD knock-in mice (which express endogenous mutant huntingtin), Cui et al
7 showed that mutant huntingtin disrupted mitochondrial function by inhibiting expression of peroxisome proliferator-activated receptor γ coactivator-1α.
Dementia is a complex neurodegenerative disorder with multiple genetic and environmental correlates. Like AT, dementia is also associated with poor DNA repair ability.
8 Recent studies have demonstrated that telomere shortening and associated DNA responses promote mitochondrial dysfunction and oxidative stress.
9 It is not clear if the reduced telomere length that is observed, is a cause or consequence of compromised mitochondrial function and oxidative stress.
10Both HD and dementia are known to have increased oxidative stress with diminished neuronal survival,
11 perhaps consequent to metabolic and mitochondrial compromise.
12,13 Because the role of mitochondrial dysfunction in HD and dementia is beginning to be understood, it was of interest to assess the loss of telomere sequence, if any, in these neurodegenerative diseases. Telomere length in peripheral blood leukocytes could be used to study these neuro-psychiatric disorders, as the leukocyte telomere length is reflective of global cellular morbidity and mortality.
9In this study, we examined the relative telomere copy/single copy gene (T/S) ratios that reflect relative telomere length in subjects with HD and dementia using Cawthon’s method.
14 We also selected patient samples of AT to measure the relative T/S ratios, as telomere attrition is well established in this syndrome.
Methods
Sample
Patients with a clinical diagnosis of HD, dementia and AT were included in the study (
Table1). The subjects in the HD group had been symptomatic for 5 years (SD, 3.3), with a mutated allele of the CAG repeats ranging from 40 to 85. The HD sample was part of a larger study of genetic and clinical correlates in HD, and details about evaluation have been reported earlier.
15The dementia subjects (N=70) were diagnosed by ICD-10 criteria and were evaluated using the Hindi Mental Status Examination scale.
16 The dementia group subjects had been symptomatic for 2.8 years (SD, 1.9), with Hindi Mental Status Examination scale scores of 11.2 (SD, 7.7). Dementia of Alzhiemer’s type formed the major group (N=57), followed by vascular dementia (N=8), fronto-temporal dementia (N=2), and Lewy body dementia (N=3).
The AT cases (N=9) were clinically diagnosed and confirmed by elevated alpha-feto protein levels. The control subjects (N=105) were corresponding age-matched subjects recruited from the general population and had no lifetime history of psychiatric illness and no known family history of neuro-psychiatric illness. Hindi Mental Status Examination scale score in control subjects for the dementia group (N=55) was 30.8 (SD, 0.7).
The study was approved by the Institutional Ethics Committee, National Institute of Mental Health and Neurosciences, Bangalore. After informed written consent, 10 mL of venous blood was collected, and DNA was isolated using the salting out method.
17 The real-time polymerase chain reaction (PCR) was setup using the Applied Biosystems 7500 system where the telomere and a single copy gene in a known quantity of DNA were amplified simultaneously and the results were recorded as number of cycles of real-time PCR required to reach threshold fluorescence (Ct or cycle threshold).
The Ct value is inversely related to the initial copy number of telomere or single copy gene. The T/S ratio (or ΔCt=Ct of telomere amplification − Ct of single copy gene amplification), which reflects number of copies of telomere per diploid genome in an assay, is an indirect measure of telomere length. The relative T/S ratio, which is calculated as the difference between ΔCt of unknown sample and normalized for ΔCt of reference DNA, enables comparison of T/S ratios across assays.
14 The following formula was used to calculate relative T/S ratios:
The reference sample was an arbitrarily chosen sample that was included in all experimental assays to permit interassay comparison. Each sample was assayed in duplicate, and Ct values that showed SD > 1 between the technical replicates were not considered for analysis. Experiments were repeated after recoding the DNA samples to eliminate sample bias.
Reproducibility of the Measured Relative T/S Ratios
Replicate assays of same sample and reference were setup at different times to calculate the interassay variation. The coefficient of variation (average standard deviation) as calculated by measuring relative T/S ratios of a sample repeated over four different assays was 6.7%. Thus, samples differing in average telomere length by as little as 13% (1.96×SD) should be distinguishable by this method at the 95% confidence interval.
14Statistical Analysis
Statistical tests were done using R software
18 and QTI plot.
19 The chi-square test was used for comparing categorical variables. The Mann-Whitney
U test (
U, two-tailed p) and Kruskal-Wallis test (χ
2, p) were used for comparison between groups of continuous variables as the relative T/S ratios and log-transformed relative T/S ratios calculated were not normally distributed and nonparametric tests are most useful for small samples.
20,21 Spearman's rank correlation (r, two-tailed p) and partial correlation (r, two-tailed p) coefficients were used for correlation analysis.
Results
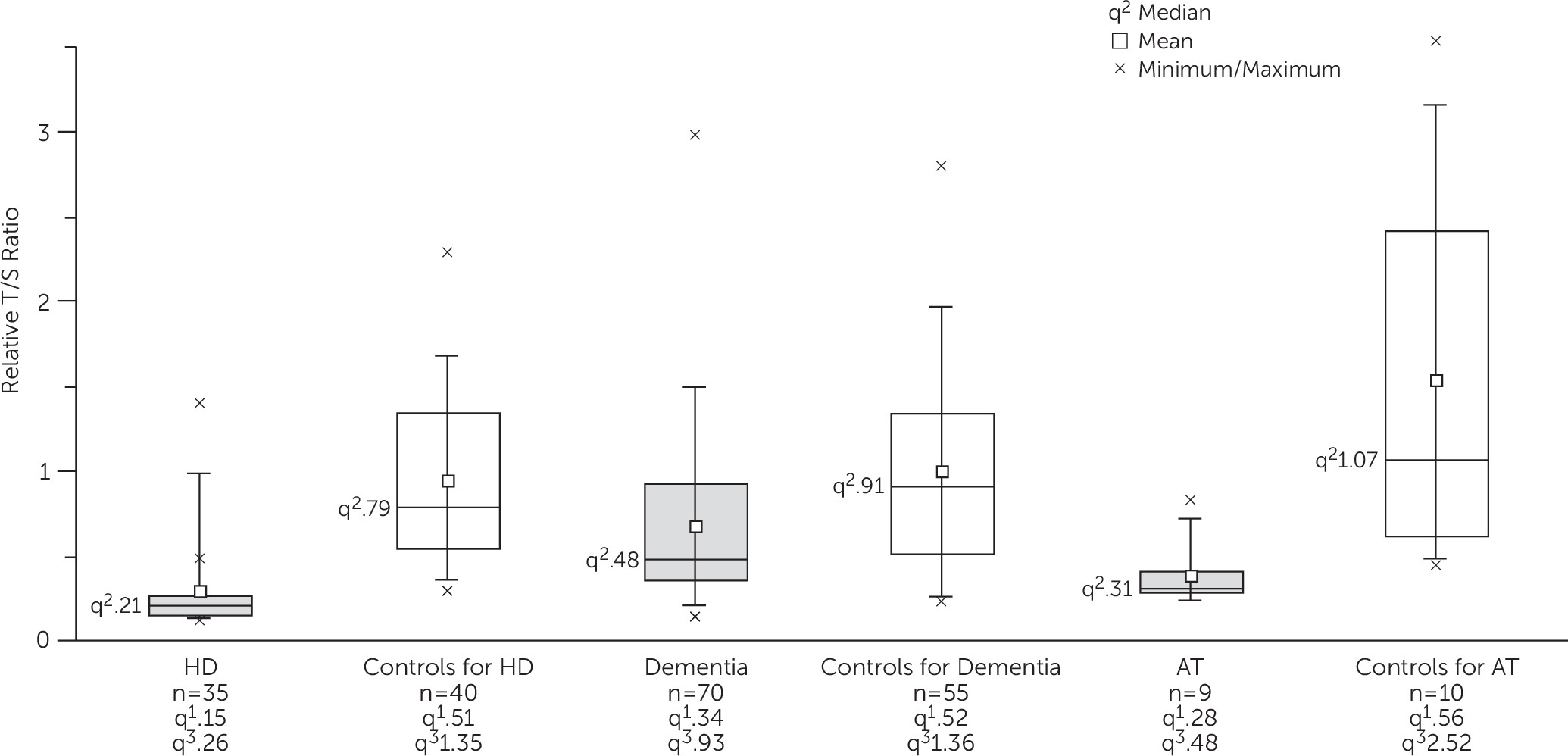
Telomere length is known to decrease with age, and because there was considerable difference in mean age between the three case groups, we used a different set of control subjects for each phenotype. There was a considerable spread of values for the relative T/S ratios within each group (
Figure 1). The relative T/S ratios were significantly lower in HD (
U = 111; p<0.0001), dementia (
U = 1211, p<0.0001), and AT (
U = 6, p=0.005) compared with the age-group and gender-matched control groups. The results remained significant after adjusting for age and gender in HD and AT groups (r=0.59, p<0.0001 and r=0.65, p=0.005, respectively) and after controlling for age at blood sampling, gender, diabetes, hypertension, and heart disease in the dementia group (r=−0.31, p=0.001) by partial correlation analysis.
The relative T/S ratios were not significantly different between genders across all groups or within individual groups (p>0.05). The relative T/S ratios did not show significant correlation between CAG repeat size (r=−0.27, p=0.18), age at onset (r=−0.05, p=0.79), and duration of illness (r=−0.22, p=0.2) in HD cases. The relative T/S ratios did not correlate significantly with age at assessment (r=0.11, p=0.36), age at onset of symptoms (r=0.1, p=0.39), and duration of illness (r=0.05, p=0.7) in subjects with dementia. The duration of illness did not correlate significantly with relative T/S ratios after controlling for age at assessment in HD (r=−0.12, p=0.48) and dementia (r=0.08, p=0.52) subjects. The relative T/S ratios did not correlate significantly with dementia subtypes (χ2 = 2.1, p=0.55) and Hindi Mental Status Examination scale scores (r=0.02, p=0.85). After controlling for age at assessment, the severity of dementia as measured by Hindi Mental Status Examination scale scores did not show significant correlation with relative T/S ratios (r=−0.02, p=0.9).
Within the control groups, children had significantly higher relative T/S ratios than adults as expected (U = 209, p=0.02). Within adult control groups, the relative T/S ratios did not differ significantly between controls for HD and controls for dementia (U = 1089, p=0.93). Our data thus confirm a significant loss of telomere sequence in the neurodegenerative disorders tested.
Discussion
The results on leukocyte telomere length in HD and dementia indicate attrition, which was also seen in the AT samples, but not in the control samples. This suggests that the observed response in lymphocytes may be shared across these syndromes.
At least four genes involved in metabolism and oxidative stress are known to be underexpressed in the striatum of patients with HD.
22 These genes are found to be down-regulated even in the peripheral leukocytes of both pre-HD carriers and patients with HD.
23 Thus, the oxidative stress known to occur in the brain is also experienced by the peripheral tissues in individuals with the mutated huntingtin gene. Sahin and DePinho
9 demonstrated that telomere dysfunction activates p53, which in turn binds and represses peroxisome proliferator-activated receptor γ coactivator-1α and peroxisome proliferator-activated receptor γ coactivator-1β promoters, thereby forging a direct link between telomere and mitochondrial biology. Soyal et al
24 recently identified alternate promoters for peroxisome proliferator-activated receptor γ coactivator-1α in HD and showed that the corresponding haplotypes influence the age of onset, suggesting that telomere homeostasis in HD may be related to peroxisome proliferator-activated receptor γ coactivator-1α function. To corroborate this finding in leukocytes, larger samples of HD can be explored for telomere attrition as a marker for neuronal pathobiology.
The lower relative T/S ratios observed in the dementia group are consistent with earlier work on telomere length in dementia.
3,25–28 Oxidative stress, aberrations in mitochondrial homeostasis, deficient DNA repair mechanisms, and decreased DNA methylation status seen in dementia have been considered as the cause for this low relative T/S ratio.
8,9,29–33Further, we did not find a significant correlation between relative telomere length and illness duration even after controlling for age at assessment in HD and dementia subjects. As this was a cross-sectional study, a longitudinal study with multiple sampling as the disease progresses would be required to understand whether the lowered relative telomere length is a cause or effect of disease progression. It is possible that the telomere attrition, which is a cellular event, could occur before the onset of symptoms of the disease. This could be assessed by measuring relative telomere length in at-risk individuals.
Induced pluripotent stem cells–derived neurons of Parkinson’s disease from carriers of single gene mutation in the leucine-rich repeat kinase 2 gene, as well as those of possible multifactorial etiology, showed similar cellular features of auto-phagosome turnover. This suggests that, in clinical syndromes arising from differing etiologies, the final cellular pathology may be shared.
34 A similar overlap of biological processes in peripheral cells in dementia and HD could be also expected. Animal studies using aged telomerase-deficient transgenic mice have shown that degenerative status, associated with short telomeres, could be reversed by activation of an inducible telomerase gene. This was evidenced by restoration of neural progenitors in the generation and maintenance of olfactory bulb interneurons in mice.
35The results demonstrate an overall loss of telomere sequence in disease. Whether the increased oxidative stress could be the cause and/or effect of telomere dysfunction needs to be explored. Methods that measure chromosome specific telomere lengths could be explored further to understand the disease specific mechanisms of telomere attrition.
In summary, we observed that leukocytes from patients with neurodegenerative disease show significant shortening of telomeres. The precise mechanism for this attrition needs to be studied further. Mutation (AT mutated/huntingtin gene)-specific mechanisms, or the complex pathways involved in dementia, can be evaluated, to help in understanding the biology of these diseases, as well as identify targets for intervention. Strategies to prevent telomere loss or increase telomere length may prevent or delay degeneration and hence the onset of symptoms in degenerative disorders. Meanwhile, the use of relative T/S ratio as a biomarker for cell senescence in diseases that involve premature aging needs to be explored further.