The link between stressful life events and the origin and development of depression has been widely investigated, providing an increasing body of evidence supporting this association.
1–3 Environmental factors likely affect individuals in somewhat different manners, therefore triggering an adaptive response to stress, which depends on both psychological and biological aspects in the interaction between stressors and individual resources. Psychological aspects include all of the cognitive processing related to incoming information; the subjective appraisal of different features related to stressors, such as magnitude and chronicity, predictability, and controllability; and potential resources to cope with them. Biological mediators include the activation of different neural structures underlying information processing, including sensory pathways, which convey environmental input to the CNS, and the resulting activation of neural and neuroendocrine cascades of molecular events, mediated by the subsequent activation of the sympathetic division of the autonomic nervous system and the hypothalamic-pituitary-adrenal (HPA) axis.
4 The efficacy of an adaptive response implies that it may be rapidly activated, to allow reacting in a successful and effective manner during stressful situations, and it should be efficiently controlled and concluded afterward. If it continues in a prolonged and excessive manner (e.g., during chronic stressful situations), it may lead to maladaptive changes, which in turn may contribute to the development of pathological conditions such as anxiety and mood disorders, including depression, particularly in individuals with increased genetic vulnerability. In this regard, various polymorphisms have been investigated as candidate genes, which are known to participate in important molecular pathways involved in the origin of depression. The presence of these genetic variations appears to be involved in the development of depression in response to stressful events, including adverse experiences during childhood and environmental stressors during adulthood.
5–10 Moreover, various studies have focused on the role of gene–environment interactions, including the search for these polymorphic variants and the role of transcriptional regulation by epigenetic mechanisms.
6,11–13 In addition, inflammatory processes associated with adaptive responses to stressful situations, with the consequent synthesis and release of proinflammatory cytokines, may lead to further maladaptive changes of neural and neuroendocrine systems, therefore contributing to the development of depressive symptoms, particularly in chronically stressed individuals.
This article aims to review the evidence for the role played by stress, associated with different converging factors, including a genetic diathesis, a history of adverse early life events, hyperactivity of the HPA axis, decreased monoamines, increased proinflammatory cytokines, and epigenetic mechanisms, such as those observed in response to environmental stressful conditions, and their potential interactions in the etiology of depression. An increased understanding of these factors and their potential interactions may lead to more effective strategies for the treatment of this disorder.
Processing of Environmental Stressors in the Brain
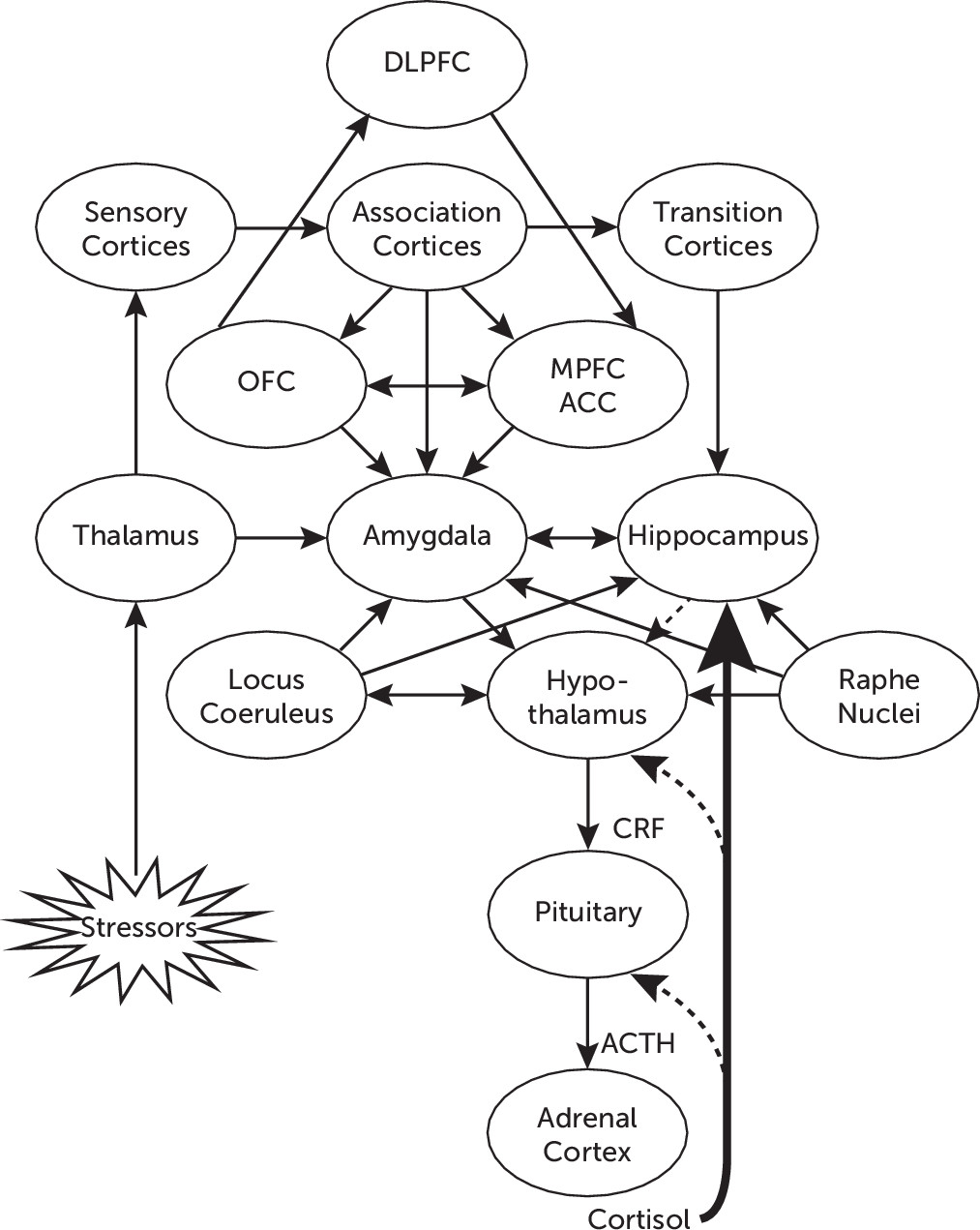
Environmental stressors are perceived and transmitted through sensory pathways to different structures in the CNS, such as the thalamus, which convey projections to the amygdala, and to sensory and association cortices, which in turn also project to different areas of the prefrontal cortex (PFC), including the orbitofrontal cortex, the medial PFC, and the anterior cingulate cortex (ACC).
4,7 Direct projections from the thalamus to the amygdala contribute to activate arousal and early alarm reactions, with the subsequent activation of the autonomic nervous system and the HPA axis, whereas indirect projections may reach the amygdala from sensory and association cortices as well as from transition cortices. The latter areas, including the entorhinal, perirhinal, and parahippocampal cortices, in turn project to the hippocampus, where sensory input is integrated with contextual cues, to convey more elaborated information to the amygdala.
14The amygdala plays a critical role in emotional processing, including the assessment of the emotional relevance of environmental stimuli as well as internal stressors. It plays a key role in the regulation of autonomic and neuroendocrine responses, through projections to the lateral hypothalamus, which mediate the activation of the sympathetic branch of the autonomic nervous system; through direct projections to the paraventricular nucleus of the hypothalamus; or indirectly through the bed nucleus of the stria terminalis, which is involved in activation of the HPA axis.
14 In addition, the amygdala shares important connections with the orbitofrontal cortex and the medial PFC,
15 including Brodmann areas 10 and 32, and the subgenual ACC (Brodmann area 25).
16 The orbitofrontal cortex (Brodmann areas 11–14) has been associated with integration of multimodal sensory stimuli and primary appraisal of their positive or negative value, therefore participating in their affective assessment.
17 The medial PFC overlaps with the ACC, particularly in the subgenual ACC,
17 which regulates emotional responses generated by the amygdala.
18 These structures are in turn connected with the dorsolateral PFC (Brodmann areas 9 and 46) and the ventrolateral PFC (Brodmann areas 45 and 47), which participate in cognitive control and voluntary regulation of emotion. The dorsolateral PFC, which has been associated with executive aspects of cognitive processing
19 (most notably with conscious processing and working memory), receives input from the amygdala through the orbitofrontal cortex and ACC.
15,17 The dorsolateral PFC reciprocally projects back to limbic structures, mostly through indirect connections to the ventromedial PFC (Brodmann area 32), which projects to the subgenual ACC.
19 It has been proposed that projections from the ventromedial PFC and the subgenual ACC exert a modulatory effect on the amygdala,
19,20 which in turn sends excitatory output to the hypothalamus,
17–19 therefore regulating the activity of the HPA axis.
Decreased volume of the subgenual ACC has been described, together with hyperactivity of the amygdala, in individuals with mood disorders,
16,21 which has been associated with the role of the subgenual ACC in the top-down regulatory pathway between the dorsolateral PFC and the amygdala, allowing conscious down-regulation of negative emotions. These corticolimbic pathways may be dysfunctional in patients with depression, in which the dorsolateral PFC, dorsomedial PFC, orbitofrontal cortex, and ACC appear to be dysfunctional, particularly during cognitive-emotional tasks, with the consequent disruption of their top-down inhibitory effect expressed in the impaired cognitive modulation of emotions.
20,21 Recovery of conscious regulation of negative emotions has been associated with clinical recovery. In addition, decreased hippocampal volume has also been observed, along with increased activity of the amygdala and reduced activity of the dorsolateral PFC.
21 More recently, we documented changes in cortical thickness in patients exposed to child abuse and neglect, with the findings specific to the nature of the abuse.
22Figure 1 illustrates the network of functional connections among different neural structures involved in adaptive responses to stress, including the processing of environmental stimuli through cortical and subcortical structures, and the activation of the HPA axis.
Role of the HPA Axis
Activation of the HPA axis is initiated in limbic structures, including direct projections from the central nucleus of the amygdala, or indirectly through the bed nucleus of the stria terminalis, which projects to the hypothalamic paraventricular nucleus, where corticotropin-releasing factor (CRF) is synthesized in parvocellular neurons and released to reach the anterior pituitary. There, CRF regulates the transcription of the proopiomelanocortin gene (a common precursor for adrenocorticotropin, β-endorphin, and related peptides) and stimulates the release of adrenocorticotropin into the systemic circulation. Adrenocorticotropin acts upon the adrenal cortex to stimulate the biosynthesis and release of glucocorticoids, particularly cortisol.
23At the molecular level, cortisol binds to mineralocorticoid receptors (type I) and glucocorticoid receptors (GRs; type II), constituting a hormone-receptor complex, which in turn undergoes conformational changes to allow its recognition and binding to a glucocorticoid response element, in the promoter region of many target genes.
24 Cortisol regulates the activity of the HPA axis through multiple negative feedback loops, which require its binding to GRs located in the paraventricular nucleus and the pituitary, where it down-regulates the synthesis and release of CRF and adrenocorticotropin, respectively, and GRs in the hippocampus, which in turn activates GABAergic projections to the paraventricular nucleus that inhibit HPA axis activity. Hence, many of the effects of cortisol may be understood as a result of transcriptional regulation of various genes, including those involved in the negative feedback loops responsible for the regulation of the HPA axis.
24In response to short-term exposure to environmental stressors, the amygdala stimulates the HPA axis with the consequent synthesis and release of cortisol,
14 which is self-regulated by negative feedback mechanisms mediated by the glucocorticoid. In addition, the HPA system interacts with CRF neurons in the amygdala, activating a positive feedback loop involved in fear and anger reactions; the HPA also activates catecholaminergic neurons, stimulating arousal and improving cognitive functions. Hence, upon exposure to acute or short-term stressors, cortisol is expected to exert widespread metabolic effects, which is mostly necessary to maintain or restore homeostasis.
25 Cortisol is actively involved in the mobilization of energetic resources, including the stimulation of gluconeogenesis with the resulting increased levels of circulating glucose, and the down-regulation of inflammatory processes, therefore contributing to coping with the stressful situation.
Chronic and persistent activation of the HPA system may disrupt physiological mechanisms, including negative feedback loops, resulting in persistent activation of the system. Circadian rhythms normally characterized by wide variations, with morning zeniths and evening nadirs, are markedly altered during chronic stress, with the consequent increase in plasma cortisol levels and blunted circadian rhythm, mostly due to increased levels of cortisol during the evening and mild changes in the morning.
25 Prolonged exposure to increased levels of cortisol may induce detrimental effects on hippocampal neurons, reducing dendritic branching and inhibiting neurogenesis.
26 Moreover, hypersecretion of CRF and cortisol was also associated with decreased hippocampal volume, particularly in individuals exposed to childhood trauma.
27 Because the hippocampus is involved in the regulation of the HPA axis, it is conceivable that patients with major depression and early life trauma who exhibit reduced hippocampal volume
28,29 may also exhibit decreased hippocampal function, therefore resulting in further sensitization of stress responses.
5 These observations support previous reports that associated the origin of depressive symptoms with decreased expression of GRs at the hypothalamic and hippocampal levels,
24 with the resulting hypercortisolism. Hence, an increasing body of evidence supports the association between chronic stress and depression at the molecular level, where hyperactivity of the HPA axis, with the consequent increase of cortisol, represents one of the most consistent findings in both syndromal mood and certain anxiety disorders.
23,26Various studies have focused on genes involved in the regulation of the HPA system, including both the mineralocorticoid receptor and GR genes, resulting in the identification of different single-nucleotide polymorphisms (SNPs). Among these, two different SNPs in the GR gene (BclI and Asp363Ser) have been associated with increased vulnerability for depression in the general population, probably through increased glucocorticoid sensitivity.
30 More recently, various studies have focused on the FK506-binding protein FKBP5, a cochaperone of hsp-90 involved in the regulation of GR sensitivity,
31 which is also involved in HPA axis responsivity. This protein is a component of the GR heterocomplex, which, upon binding of cortisol, is replaced by FKBP4, which in turn facilitates the nuclear translocation of the hormone-receptor complex and its transcriptional activity.
32 Altered GR function may lead to impaired feedback regulation, with the resulting HPA hyperactivation commonly observed in chronic stress and depression. Therefore, various SNPs have been identified in the FKBP5 gene, some of them associated with increased FKBP5 protein expression, which in turn may lead to changes in GR, with the resulting effect on HPA axis regulation.
32 Increased FKBP5 protein expression may reduce hormone-binding affinity and may interfere with the translocation of the hormone-receptor complex. It is noteworthy that glucocorticoids may induce increased expression of this cochaperone, constituting an intracellular negative feedback loop to regulate GR activity.
33 One of the SNPs of the FBPP5 gene, defined as the substitution of a cytosine (C) by a thymine (T) and therefore identified as the high-induction allele T, was associated with increased FKBP5 protein expression and altered HPA response. Upon exposure to stressful stimuli, carriers of the T allele exhibited slower recovery of the cortisol response and homozygous carriers of the allele who experienced severe abuse during childhood presented increased vulnerability for the development of depression during adulthood,
34 which may also be associated with having an increased number of depressive episodes.
32Role of CRF
CRF-containing circuits in the CNS play a critical role in the coordination of the stress response, both as a neuroendocrine factor regulating the HPA axis and through its function as a neurotransmitter, mediating behavioral, immune, and autonomic responses to stress.
35 CRF neurons are localized throughout different cortical areas, participating in neural pathways involved in cognitive responses, and limbic areas such as the central nucleus of the amygdala and the bed nucleus of the stria terminalis, where it participates in the regulation of emotional responses.
23 CRF projections from the amygdala have been shown to reach the hypothalamic paraventricular nucleus (therefore enhancing the activation of the HPA axis in response to stress) and the monoaminergic nuclei in the brainstem, including the locus coeruleus (LC) and the raphe nuclei (RN).
3 Moreover, CRF stimulates norepinephrine release in the LC,
36 with the consequent noradrenergic activation of the autonomic nervous system and the HPA axis, while mainly inhibiting serotonergic neurons in the RN,
37 which in turn may affect other structures through serotonergic projections to the amygdala, hippocampus, and paraventricular nucleus.
3 Therefore, through the regulation of these monoaminergic systems, CRF participates in neurobiological processes underlying mood and anxiety disorders, producing anxiogenic and depressogenic effects.
35 Increased CSF concentrations of CRF have consistently been reported in depressed and suicidal patients.
38 In addition, CRF may also be involved in anxiety and the encoding of emotional memories,
23,35 playing a critical role in the stress response not only during adulthood but also in mediation of the long-lasting effects of trauma and other early life stressful experiences. Moreover, increased levels of CRF may also be involved in neuroplastic changes induced by chronic stress,
39 and this effect may also be enhanced by glucocorticoids as a component of the stress response.
40Various studies have focused on CRF, CRF-binding protein, and CRF type 1 receptor (CRHR1) genes, resulting in several important findings.
41 Indeed, several SNPs in the CRHR1 and haplotypes formed by certain SNPs involved in mediating the effects of early adverse experiences on the risk for adult depression have been identified.
42 Upon binding to CRF, this receptor participates in the activation of the HPA axis and plays a critical role in emotional and cognitive functions mediated by CRF in extrahypothalamic brain regions, including the amygdala and the LC,
35 therefore influencing arousal, attention, conscious perception of emotional experiences, and memory consolidation. Two haplotypes formed by different SNPs in the CRHR1 gene were associated with reduced symptoms of depression in subjects exposed to early stressful experiences. Because CRHR1 may be critically involved in the consolidation of emotionally charged memories, such as those produced by childhood aversive experiences, it was proposed that carriers of two copies of these haplotypes, which also exhibited overrepresentation of the protective alleles of the studied SNPs,
42 may have altered activation of memory consolidation processes. This may lead to decreased emotional influence in the cognitive processing of these memories, therefore protecting the individual from his or her potentially depressogenic and anxiogenic effects.
43Role of Serotonin
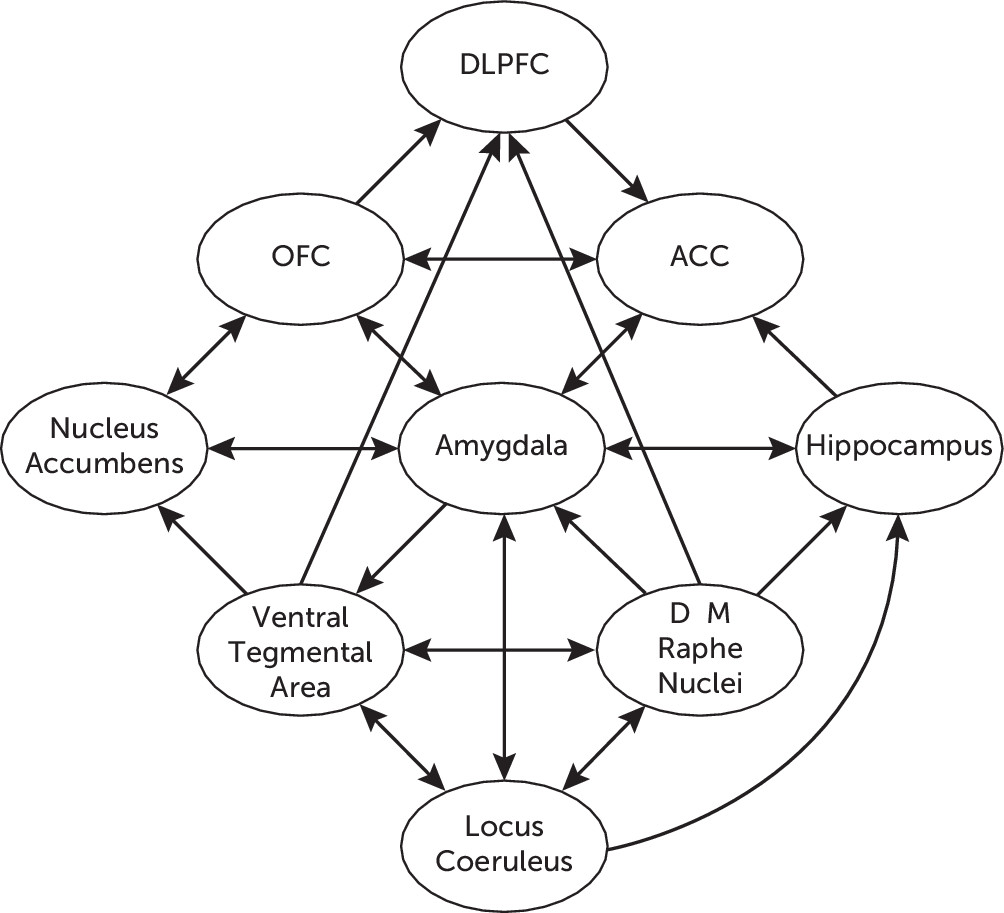
The serotonergic hypothesis of depression posits deficient serotonergic activity in the CNS with increased vulnerability for the development of depression. The main groups of serotonergic neurons in the CNS are located within the boundaries of the RN, where an array of ascending projections arise from the dorsal RN (B6 and B7) and the medial RN (B8). The dorsal RN–forebrain tract projects to the PFC, amygdala, nucleus accumbens, and ventral hippocampus, among other forebrain structures,
44 and it participates in the state of anticipatory anxiety and thus plays an adaptive role during stressful situations.
45 The dorsal RN–forebrain tract has been associated with activation of the limbic structures (e.g., the amygdala) in the presence of environmental stressors associated with unpleasant experiences, and it is also involved in the regulation of potential emotional reactions. Alterations of this system, particularly involving dorsal RN–amygdala projections, may be associated with symptoms of anxiety.
45 The medial RN–forebrain tract projects to the dorsal hippocampus and hypothalamus, among other neural structures,
44,45 and it participates in conferring tolerance to unpleasant, unavoidable, and persistent aversive stimuli such as those perceived during chronic stressful situations. The medial RN–forebrain tract is also associated with adaptive control on negative emotional experiences. Therefore, alterations of this system, particularly involving medial RN–hippocampal projections, may be associated with decreased tolerance to aversive stimuli, learned helplessness, and subsequent depression.
45,46 Serotonergic neurons in the RN are also interconnected and are physiologically integrated with other monoaminergic systems in the brainstem, including noradrenergic and dopaminergic circuits.
47 It has been shown that both the dorsal RN and the medial RN receive noradrenergic projections,
48 which appear to be excitatory. The LC receives serotonergic projections from the RN reciprocally,
48 which appear to exert an indirect modulatory effect by inhibiting glutamatergic activation of the LC. The dorsal RN also modulates dopaminergic activity through projections to the ventral tegmental area, which appear to be excitatory,
49 and dopaminergic projections to the dorsal RN reciprocally exert an indirect inhibitory effect by increasing the activity of somatodendritic 5-hydroxytryptamine (serotonin [5-HT]) autoreceptors.
44Figure 2 illustrates the network of functional connections between different neurotransmitter systems in the CNS, as well as their respective connections with different cortical and limbic structures involved in the stress response.
At the molecular level, 5-HT is released into the synaptic cleft, where it binds to both presynaptic and postsynaptic receptors. A growing number of 5-HT receptors have been identified, including 14 different types, classified in seven families with various subtypes each. Each of the serotonin receptor subtypes exhibits a unique regional neuroanatomic distribution, conferring specificity on the effects of activation of this widespread and diffuse serotonergic innervation. Synaptic concentrations of 5-HT are regulated by the serotonin transporter (5-HTT), which is responsible for its reuptake, therefore regulating its availability to bind and activate specific 5-HT receptors.
47 The 5-HTT is believed to be the primary molecular target of selective serotonin reuptake inhibitors antidepressants. Hence, 5-HTT blockade by selective serotonin reuptake inhibitors is translated into higher 5-HT concentrations in the synaptic cleft, allowing increased activation of 5-HT receptors.
46,47 The clinical efficacy of antidepressants is not directly associated with this acute mechanism; instead, it is linked to more adaptive changes. Continuous administration of selective serotonin reuptake inhibitors leads to desensitization or down-regulation of somatodendritic 5-HT
1A autoreceptors in the RN after several days (which are known to moderate the release of 5-HT into the synaptic cleft) and up-regulation of postsynaptic 5-HT
1A and desensitization of 5-HT
2A receptors.
50In addition to serotonergic projections directly involved in cognitive and emotional functions, projections from the RN have been shown to innervate CRF-containing neurons in the paraventricular nucleus.
51 There is evidence that these projections stimulate the HPA axis and the autonomic nervous system; glucocorticoids and catecholamines may reciprocally affect the serotonergic system during stressful situations.
46 Various studies have shown that postsynaptic 5-HT
1A receptors in different limbic structures may be down-regulated or desensitized by glucocorticoids or exposure to chronic stress.
52,53 In addition, it has been shown that cortisol may increase 5-HT uptake in vitro, an effect attributed to increased expression of the 5-HTT gene by the glucocorticoid,
54 therefore providing further support for the reciprocal regulation of the HPA and 5-HT systems and their potential interplay in the interface between stress and depression.
46Various studies have also focused on the structure of the 5-HTT gene, in which a polymorphism was identified in its promoter region.
55 The promoter activity is regulated by sequence elements located in the upstream regulatory region, known as the 5-HTT gene-linked polymorphic region (5-HTTLPR), where a short (S) and a long (L) allele have been identified.
6 Hence, the short promoter variant (5-HTTLPR-S) was associated with decreased transcriptional efficiency compared with the long allele (5-HTTLPR-L), resulting in decreased expression of the 5-HTT gene,
55 which may affect the modulation of serotonergic activity in response to stress. This notion has been supported by multiple clinical and preclinical studies,
56 including evidence observed in functional brain imaging studies, in which carriers of the S allele (homozygous or heterozygous for the short allele) exhibited increased amygdala reactivity to fearful and threatening stressors compared with those homozygous for the L allele,
57 which suggests that variations in the 5-HTT gene may be involved in psychological responses to stress.
6 Although various studies have shown increasing evidence that this polymorphism moderates the relationship between stress and depression,
56 there are still other studies suggesting certain controversy around this hypothesis.
The amygdala participates in the regulation of emotional reactions to stressful events, and its increased reactivity was associated with anxiety and altered mood regulation.
14 Hence, a potential association between 5-HTT gene polymorphism and increased reactivity of the amygdala in response to negative stressors
58 may contribute to a better understanding of the potential effect of the molecular mechanisms underlying this association. Moreover, the amygdala also plays a critical role in the activation of the HPA axis, and hyperactivation of the amygdala may also lead to increased plasma levels of cortisol. Indeed, carriers of the S allele exhibit increased activation of the amygdala and elevated cortisol levels in response to a laboratory stressor.
11 The association between the 5-HTTLPR-S variation and a potentially decreased expression of the 5-HTT gene may appear paradoxical, considering the potential vulnerability attributed to 5-HTTLPR-S carriers. Therefore, it is conceivable that alterations in 5-HTT gene regulation (and consequent effects on synaptic 5-HT levels) may differ, with the former expressed as a result of constitutive conditions and the latter triggered by environmental factors. It has been proposed that 5-HTTLPR-S carriers may exhibit “essentially” increased concentrations of 5-HT, which may result in down-regulation of postsynaptic 5-HT receptors. This may lead to a relative desensitization of the serotonergic system,
58 providing a potential explanation for the vulnerability exhibited by 5-HTTLPR-S carriers. By contrast, up-regulation of the 5-HTT gene, associated with the effect of environmental stressors and the resulting hyperactivation of the HPA axis and hypercortisolism, may lead to increased 5-HT reuptake and decreased concentrations of 5-HT in the synaptic cleft,
54 which has been widely associated with the development of mood disorders.
Role of Dopamine
Dopamine has also been implicated in the neural mechanisms of stress responses, including stress-related regulation of the HPA axis, as well as in the pathophysiology of depression.
59,60 The main groups of dopaminergic neurons in the CNS comprise the retro-rubro field (A8), the substantia nigra pars compacta (A9), and the ventral tegmental area (A10), where the mesolimbic and mesocortical pathways arise. The mesolimbic pathway projects mainly to the nucleus accumbens and other limbic structures, including the amygdala, hippocampus, bed nucleus of the stria terminalis, and septum. This pathway is implicated in the processing and reinforcement of rewarding stimuli, motivation, and the subjective experience of pleasure.
59 The mesocortical pathway projects mainly to the PFC, ACC, and entorhinal cortex and is critically involved in cognitive functions such as concentration and working memory.
59Environmental stressors provoke increased activity in the amygdala, which in turn may increase the concentrations of dopamine in the mesocortical pathway (particularly in the PFC), therefore conferring exaggerated salience to relatively mild negative stimuli
60 and contributing to the resulting negative bias in cognitive processing. Regarding the mesolimbic pathway, it has been shown that stressful events may induce opposite responses, depending on the potential controllability of the stimuli,
61 and the consequent subjective assessment. Therefore, exposure to acute and controllable stressors was associated with increased dopamine release in the ventral striatum, whereas exposure to chronic and uncontrollable stressful stimuli was associated with decreased dopaminergic activity
61 with resulting anhedonia. Moreover, it has been shown that unavoidable or uncontrollable stressors may lead to decreased dopamine release in the nucleus accumbens and impaired response to environmental stimuli, which may result in the expression and exacerbation of depressive symptoms induced by stress.
62 The inability to experience pleasure, associated with loss of interest and motivation in usual activities, constitutes the pathognomonic anhedonia exhibited by patients with depression,
59,60 and it has been shown that impaired dopaminergic function is critically involved in altered reward processing underlying anhedonia.
63,64 Moreover, the mesolimbic dopaminergic pathway, particularly the nucleus accumbens, participates in the processing of rewarding and hedonic experiences in association with the orbitofrontal cortex, which may be involved in the subjective assessments of hedonic and rewarding value.
65 The orbitofrontal cortex is connected with the ACC and dorsolateral PFC, where this emotional input participates in cognitive processes; by contrast, the nucleus accumbens receives dopaminergic projections from the ventral tegmental area, which may be enhanced by glutamatergic stimulation from the amygdala, to increase motivation.
65 Substantial interaction has also been described between the ventral tegmental area and the RN,
59 which may be critically involved in emotional processing.
Because increased dopamine release in the mesolimbic pathway has been observed not only in response to rewarding stimuli but also in the presence of aversive situations (particularly when these are perceived as controllable and escapable
61), it has been suggested that dopamine plays an adaptive role associated with motivation, increased arousal, and behavioral control in response to stress, including both appetitive and aversive conditions.
66Role of Norepinephrine
Catecholamines (and more specifically norepinephrine) have long been posited to play a major role in the pathophysiology of affective disorders, forming the catecholamine hypothesis of depression. The main group of norepinephrine-containing neurons in the CNS is located within the LC (A6), where various projections arise to widely innervate cortical and subcortical areas,
48 including the amygdala, the hippocampus, and the paraventricular nucleus of the hypothalamus.
36 Projections from the LC to the ventral tegmental area have been described, in which norepinephrine has been shown to potentiate dopamine release. Projections from the LC to the RN have also been described, in which norepinephrine exerts regulatory effects on 5-HT release.
48 There is also evidence of reciprocal regulation between norepinephrine and 5-HT, not only through connections between both aminergic systems but also through limbic structures such as the hippocampus.
67 In addition, reciprocal connections between norepinephrine - and CRF-containing neurons suggest a critical role of the LC in the regulation of neural and neuroendocrine responses to stress.
36In response to acute stressors, norepinephrine is released throughout different structures in the CNS, resulting in enhanced arousal and hypervigilance, in the context of adaptive responses to stress. Moreover, activation of the LC has been associated with subsequent stimulation of the lateral hypothalamus, which in turn participates in the activation of the sympathetic branch of the autonomic nervous system, therefore complementing the adaptive response to stress.
36 A potential dysfunction of the LC has been observed during chronic stress (particularly upon exposure to unavoidable or uncontrollable stressors), leading to altered norepinephrine release, which was associated with some features of learned helplessness as well as problems in cognitive functions such as attention and memory, which are frequently observed in depression. In addition, dysregulation of the norepinephrine system has also been described in altered states of arousal,
48 which is commonly observed in anxiety disorders as well as in depression.
Neuroplasticity and Neurogenesis: Role of Neurotrophic Factors
Several studies have focused on the potential role of neurotrophic factors in critical neural processes, with particular attention on the neurotrophin family, which is composed of nerve growth factor, brain-derived neurotrophic factor (BDNF), neurotrophin-3, neurotrophin-4/5, and neurotrophin-6. Among these neurotrophins, a growing body of research has focused on the role of BDNF in the regulation of brain development, neuroplasticity, and neurogenesis.
68 Various studies strongly suggest that decreased levels of BDNF may lead to depressive symptoms, whereas up-regulation of BDNF is associated with clinical recovery.
69 In vitro studies have demonstrated that BDNF may decrease 5-HT uptake, suggesting a potential role of the neurotrophin in regulation of 5-HTT.
70 Chronic stress, with the resulting activation of the HPA axis, may damage neurons in certain CNS structures (particularly in the hippocampus, where high levels of GRs have been found) and these changes have been associated with decreased availability of neurotrophic factors such as BDNF.
71 Moreover, it has been shown that increased levels of glucocorticoids, at least partially, may be involved in down-regulation of BDNF.
72 By contrast, it has been demonstrated that various antidepressants increase the expression of BDNF in the hippocampus
69 in a dose-dependent and time-dependent manner, which is consistent with the time dependency of therapeutic effects of antidepressants, therefore suggesting a role for BDNF in their mechanism of action.
73 The potential association between successful pharmacotherapy and the observed up-regulation of BDNF in the hippocampus suggests that BDNF may be involved in the long-lasting effects of antidepressants through neuroplastic changes in certain neural structures such as the hippocampus, amygdala, and PFC.
69 Moreover, it has been shown that BDNF and 5-HT may induce hippocampal neurogenesis.
74Most neurons in the CNS are generated during early periods of development, although more recent studies have demonstrated that some neural structures, such as the dentate gyrus of the hippocampus, actually continue generating neurons later in life.
75 Therefore, neurogenesis in the adult CNS may be stimulated by special conditions, particularly those related to enhanced hippocampal activity and increased levels of 5-HT,
76,77 but it may be inhibited by stressful situations and increased levels of glucocorticoids.
78 Under chronic stress conditions, with increased activation of the HPA axis, inhibition of hippocampal neurogenesis may interfere with the formation of new cognitions, therefore contributing to provoking and sustaining ongoing depressogenic conditions. According to this hypothesis, successful therapeutic interventions may require recovery of the normal rate of hippocampal neurogenesis. This recovery may be associated with a direct effect of antidepressants through increasing levels of 5-HT
75 or indirectly through modulation of the HPA axis and increasing levels of BDNF, which was associated with up-regulation of neuroplasticity and increasing neurogenesis. This hypothesis remains quite controversial because of failure to confirm the increase in neurogenesis after long-term antidepressant treatment.
79Various studies have focused on BDNF gene regulation and variations potentially involved in mood disorders, resulting in the identification of different SNPs. Among these, an SNP has been identified at nucleotide position 196 in the coding region of the BDNF gene, where a guanine (G) is replaced by an adenine (A), resulting in the substitution of valine (Val) by methionine (Met) at codon 66, which is thus termed Val66Met. This is where the presence of a Met allele has been associated with a functional alteration (i.e., abnormal intracellular trafficking and decreased secretion of BDNF).
73,76 Studies on carriers of the Met-BDNF allele revealed relatively smaller hippocampal volumes compared with those individuals who were homozygous for the Val-BDNF allele.
73 This was also associated with reduced hippocampal activation and deficient cognitive performance,
12,73 which have also been associated with lower emotional stability and increased vulnerability for the development of depressive symptoms.
Inflammatory Processes: Role of Cytokines
It has been demonstrated that acute and chronic psychosocial stress may activate inflammatory responses.
80 Increased blood concentrations of proinflammatory cytokines, such as interleukin-1, interleukin-6, and tumor necrosis factor-alpha, have been associated with the effect of diverse environmental stimuli, including psychosocial stress,
81 and this immune activation has also been observed in major depression.
82 Moreover, major depression may induce increased inflammatory responses to stress, and this has been observed mostly in patients exposed to adverse early life events, therefore suggesting a link between these and increased inflammatory responses to stress later in life.
80 To understand the role of proinflammatory cytokines in chronic stress and the subsequent development of depression, various studies have focused on their potential mechanisms of action. Environmental stressors activate the sympathetic branch of the autonomic nervous system, with the resulting release of catecholamines, which in turn activates their receptors on immune cells and thus stimulates the release of proinflammatory cytokines.
83 Chronic inflammatory responses in the CNS may result in excessive release of proinflammatory cytokines, which in turn may lead to decreased concentrations of neurotrophins (including BDNF), leading to impaired neuroplasticity
83 and decreased neurogenesis (particularly in the hippocampus
82), which have been associated with the origin of cognitive impairment and mood disorders. Proinflammatory cytokines have also been involved in regulation of the HPA axis, stimulating release of CRF with resulting hypercortisolism,
83 which has been associated with reduced sensitivity of GRs and glucocorticoid resistance.
81,83 Increased levels of cortisol, such as those observed during chronic stress, may lead to decreased synthesis of 5-HT due to reduced activity of the rate-limiting enzyme tryptophan hydroxylase. Hypercortisolism has been also associated with increased activity of tryptophan dioxygenase (indoleamine-pyrrole 2,3-dioxygenase), which is responsible for the degradation of tryptophan to kynurenine, with the resulting decreased synthesis and release of 5-HT.
83 Proinflammatory cytokines such as interferon have also been involved in the modulation of this pathway, stimulating indoleamine-pyrrole 2,3-dioxygenase and thus leading to reduced synthesis of 5-HT and increased synthesis of kynurenine.
84 Degradation of kynurenine leads to the formation of 3-hydroxykynurenine, which produces free radical species involved in oxidative stress, and kynurenic acid and quinolinic acid, which activate the glutamatergic system. This leads to neurotoxicity and neuronal apoptosis, which are also involved in the pathophysiology of depression.
83,84 In addition, certain proinflammatory cytokines, such as interleukin-1 and tumor necrosis factor, have been shown to affect serotonergic neurotransmission by stimulating the 5-HTT and thus reducing intersynaptic concentrations of 5-HT in the CNS.
83,85Understanding the molecular mechanisms underlying neuroinflammatory processes in the CNS, particularly the role played by proinflammatory cytokines in mood disorders, has inspired various studies aimed at improving depressive symptoms by attenuating these processes. Preclinical studies have demonstrated the efficacy of certain anti-inflammatory cytokines to block the depressive-like state induced by proinflammatory cytokines in rodents.
83 Other studies have also approached the consequences of proinflammatory cytokines, antagonizing the activity of the glutamatergic system, activated by the kynurenine pathway.
81Stress, Appraisal, and Coping: Role of Psychological Vulnerability
Psychological vulnerability depends on various features related to stressful life events (including strength, intensity, and length of the impact) and the availability of personal resources to cope with them. More remarkably, however, it may depend on cognitive appraisal, particularly the balance between stressors and individual resources, and the resulting coping strategies.
86 Chronic exposure to unavoidable and uncontrollable stressors may lead to decreasing cognitive and behavioral coping strategies to handle environmental events, mostly as a result of cognitive appraisals that personal resources are not enough, which has been associated with increasing feelings of helplessness.
86 According to the cognitive model of depression,
87 early life experiences provide the background to develop cognitive schemas, which in turn represent the basis to transform simple data into cognitions that are learned and stored in long-term memory. Adverse early life events, including childhood sexual or physical abuse
88 and peer victimization
89 (also known as bullying), may contribute to the formation of particular cognitive schemas. These schemas may be inactive during long periods and reactivated by new experiences at a later time, particularly those with strong emotional valence. In response to stressful situations in adulthood, activated dysfunctional schemas may induce negative biases during information processing, with consequent dysfunctional effects, including cognitive processing, emotional reactions, and behavioral responses, constituting the essential core of cognitive vulnerability.
87 Therefore, dysfunctional schemas shaped during childhood, with systematic negative biases, may lead to negatively biased appraisals, with consequent limitations in further processing of the resulting cognitions, therefore leading to feelings of helplessness and subsequent depression.
Epigenetics: Role of Gene–Environment Interactions
The term
epigenetics refers to heritable characteristics that are not determined by structural changes in the underlying genetic sequence. At the molecular level, epigenetic mechanisms involve biochemical changes of nucleotides, without altering the DNA sequence, and the associated histone proteins, which constitute chromatin. Changes in the structure of chromatin may affect gene expression by allowing transcription factors to gain access to gene regulatory elements. Hence, environmental factors may induce changes in the chromatin state, which in turn may improve exposure of genes to the impact of different transcription factors, therefore increasing or decreasing gene expression while the original DNA sequence remains unaltered.
90 Potential changes include DNA methylation, which has been associated with down-regulation of gene expression; histone acetylation, which may induce up-regulation of gene expression; and histone methylation and phosphorylation, both of which may lead to activation or repression of transcriptional events.
90 Recent research has contributed to identifying epigenetic mechanisms in the context of stressful situations, which may induce long-lasting changes in gene expression in different neural structures. In turn, such changes have been associated with the development of stress-related conditions such as anxiety disorders and depression. Preclinical studies have revealed that chronic stress may regulate histone acetylation in the hippocampus, inducing transient increases and subsequent decreases; transient increases have also been observed in the amygdala.
91In addition, preclinical studies also revealed that increased levels of CRF, observed during chronic stress conditions, have been associated with decreased DNA methylation at the promoter region of the CRF gene.
92 Moreover, a history of early adverse experiences has been associated with changes in histone markers and DNA methylation of the GR gene, particularly in the hippocampus, and changes in DNA methylation have also been observed in the GR and BDNF genes.
41 Therefore, chronic stress, including early stressful experiences, may induce diverse epigenetic changes in different neural structures, with a subsequent effect on their respective functions. This, in turn, may predispose individuals to increased vulnerability to stress and to the development of diverse clinical conditions such as depression.
Childhood Trauma: Role of Early Adverse Experiences
Early life stress, defined as adverse conditions and traumatic events experienced during childhood, represents a major factor of vulnerability in the origin and development of depression and bipolar disorder.
3,5,27 The association between a history of adverse and traumatic experiences during childhood and the development of mood disorders later in life has been observed particularly after additional stressful events during adulthood.
5 It has been shown that adverse early life events (including abuse, neglect, or loss) contribute to the formation of dysfunctional cognitive schemas, which may induce negative biases in response to stressful situations at a later time, therefore contributing to generating cognitive vulnerability.
56 This mechanism was also recently described in victims of bullying.
89 Moreover, it has been proposed that certain early life events, such as neglect, may lead to the formation of dysfunctional attitudes; this has also been associated with long-term hyperactivity of the HPA axis.
93 The effect of adverse early life events has been conclusively demonstrated to induce long-lasting changes in neural and neuroendocrine systems involved in adaptive responses to stress, particularly in CRF neurotransmission.
23 This, in turn, may be translated into persistent sensitization and increased responsiveness to stress.
3,5 Increased levels of CRF may lead to hyperactivity of the HPA axis and hypercortisolism, which may induce morphologic changes such as reduced hippocampal volume.
72 In this regard, various studies have focused on the role of hippocampal GRs, and increased levels of cortisol (in a sustained and prolonged manner) have been shown to induce down-regulation of GRs in certain areas of the hippocampus.
94 Moreover, additional research has suggested that the availability and efficacy of hippocampal GRs may be permanently affected as a result of early stressful experiences,
88 therefore contributing to glucocorticoid resistance and the consequent hyperreactivity of the HPA axis observed in response to additional stressful situations. In addition, increased concentrations of cortisol and decreased GR availability, induced by stressful situations during childhood, have been associated with decreased hippocampal volume and neural activity in adulthood as well as increased reactivity of the HPA axis, with the consequent functional alterations observed in adulthood.
88,95 A history of early life adverse experiences was also associated with hyperreactivity of neural and neuroendocrine responses to stress, which is reflected through increased CRF activity, hypercortisolism, and glucocorticoid resistance.
27,96Klengel et al.
97 reported that a polymorphism in the FKBP5 gene increases the risk for the development of stress-related psychiatric disorders in adults by an allelic-specific, child abuse/neglect–dependent DNA demethylation in functional glucocorticoid response elements of FKBP5. Thus, activation of a sensitized system in the presence of additional stressful situations later in life may result in an exaggerated and maladaptive activation of the stress response, therefore generating increased vulnerability to the development of depressive symptoms upon exposure to additional stressors in adulthood.
42,98Conclusions
The role of stressful life events in the origin and development of depression may be conceptualized as the result of multiple interactions between the effect of environmental stressors and individual factors of vulnerability.
Figure 3 illustrates the role of these factors and their potential interactions at the interface between chronic stress and depression. Genetic factors, including SNPs, may be associated with functional and structural alterations in certain neural structures, including increased reactivity of the amygdala and decreased function of the hippocampus. Adverse early life events have been shown to engender biological changes in the developing CNS, as well as psychological changes reflected in the formation of dysfunctional cognitive schemas,
99 with the resulting biased cognitive processing of environmental stimuli, which may be translated into cognitive and emotional vulnerability.
87 These may be further activated in response to stress in adulthood, contributing to increased vulnerability to depression.
27The impact of abuse and neglect during childhood clearly leads to persistent changes in neural and neuroendocrine systems involved in the regulation of adaptive responses. Functional or structural alterations in the CNS, particularly in the cerebrocortical regions as well as in the amygdala and the hippocampus, along with cognitive biases, may induce biological changes such as increased levels of CRF. Upon exposure to environmental stressors, this mechanism may be translated into hyperactivity of the HPA system, with increased levels of CRF and cortisol, which in turn may lead to transcriptional events. Such molecular changes affecting different aminergic systems, particularly on the regulation of 5-HT together with altered cognitive processing, may result in emotional changes, thereby predisposing to symptoms of anxiety and depression. Therefore, multiple vulnerability factors (including psychological, biological, cognitive, genetic, and epigenetic factors) converge on different aspects of HPA regulation. This complex set of pathways likely links vulnerability to stress with the pathogenesis of depression. In addition, environmental stress has also been associated with inflammatory responses in the CNS with excessive release of proinflammatory cytokines, which may lead to further stimulation of the HPA axis, with resulting hypercortisolism and impaired 5-HT neurotransmission. Proinflammatory cytokines have also been associated with decreased neurotrophins, with resulting decreases in neuroplasticity and neurogenesis. Therefore, a better understanding of the molecular mechanisms underlying these processes may allow novel strategies aimed at improving depressive symptoms by attenuating neuroinflammation.
The observation that some individuals may exhibit stronger vulnerability to environmental stressors but others may be less sensitive, more resistant, or even resilient to similar experiences highlights the importance of further investigation of the nature of different risk factors. Future research should focus on further understanding the neurobiological background underlying these factors and should identify potential windows of intervention, including neural and molecular mechanisms involved in the interface between cognitive processing of environmental stressors and their potential effects in epigenetic processes. This may lead to the development of more successful treatments aimed at not only restoring altered neural and neuroendocrine mechanisms but also preventing the development of anxiety and mood disorders in vulnerable individuals.
This may be achieved either by identifying different vulnerability factors, which in turn may become targets for novel therapeutic interventions, or by increasing and promoting protective resources in individuals exposed to stressful conditions, particularly those exposed to traumatic events or adverse conditions during childhood.