Robert Gaupp, a disciple of Emil Kraepelin, was among the first to systematically describe forms of depression related to “arteriosclerotic brain disease,” such as mood disorders with cognitive and motor deficits caused by cortical strokes (for further details, see supplementary reference [SR] 1 in the
online supplement). Gaupp subsequently described subtypes of cerebrovascular emotional disorders, such as anxious depression progressing onto “mental weakness, affective lability, sentimentalism and euphoria”; short-lived anxious agitation with… “shouting and crying [leading] to arteriosclerotic lethargy”; aphasia comorbid with “anxious delusional depressive agitation”; “melancholia-like depression with delusions of unworthiness and self-reproach”; dysphoric hypochondriasis with anxiety and preserved insight; and melancholia followed by nocturnal anxiety and panic attacks (SR1,
online supplement). Later, Sir Martin Roth suggested an association between atherosclerotic disease and depression, and Folstein et al. demonstrated that depression was significantly more common among patients with stroke than among patients with comparable physical impairments caused by orthopedic injuries (
1) (SR2,
online supplement).
The literature on poststroke depression (PSD) has grown exponentially over the past four decades. In the present review, we address relevant conceptual issues in the diagnosis of PSD, as well as its prevalence, risk factors, relationship to physical and cognitive impairment, and association with mortality. We also review treatment and prevention of PSD, its etiology, and suggestions for future research.
Diagnosis of PSD
DSM-5 stresses the difficulty of diagnosing major depression among individuals with a general medical condition and suggests that depressive symptoms should count toward a diagnosis of major depression “except when they are clearly and fully attributable to a general medical condition” (
2). Thus, the question arises of how to diagnose depression when stroke symptoms overlap with the criteria for major depression (e.g., weight loss, fatigue, and sleep changes). One option is to use “nonvegetative symptoms,” such as dysphoria, anhedonia, guilt or worthlessness, impaired concentration or indecision, and suicidal thoughts; however, DSM-5 states that use of the full criteria for major depression and the use of only nonvegetative symptoms may identify the same individuals (
2).
One strategy to diagnose PSD is to divide symptoms of depression into somatic (vegetative) and psychological categories. Whereas this classification may have heuristic value, the delimitation of these putative domains lacks consistency and validity, and symptoms may overlap (e.g., fatigue may be considered either a somatic or psychological symptom). Cohen-Cole and Stoudemire suggested four different strategies to diagnose depression among the physically ill (
3). The “inclusive” strategy counts depressive symptoms regardless of whether they may have been produced by the physical illness. The “etiological” strategy counts symptoms only if the examiner considers them causally related to the disease. The “substitutive” strategy replaces somatic symptoms of depression with psychological ones, and the “exclusive” strategy removes symptoms from the diagnostic criteria if they are not more frequent among patients with depression than among those without depression. We will discuss the abundant empirical evidence that supports the inclusive strategy. De Coster et al. reported good sensitivity for the diagnosis of PSD with somatic symptoms, such as decreased appetite, psychomotor retardation, and fatigue, thus arguing against the substitutive or exclusive diagnostic approaches (
4). Several studies have compared the phenomenology of depression in stroke to the phenomenology of depression associated with other acute medical conditions. Lipsey et al. and Andersen found no significant differences in symptoms of depression between patients with PSD compared with patients with primary or functional (i.e., no known brain injury) depression (
5) (SR3,
online supplement). Paradiso and Robinson found a significant increase in both somatic and psychological symptoms of depression among patients with PSD compared with a control group of stroke patients without depression (
6). When somatic symptoms of depression were excluded, the frequency of a diagnosis of PSD remained unchanged. Fedoroff et al. reported similar findings, concluding that both somatic and psychological symptoms of depression are strongly associated with sad mood after stroke (
7). Cumming et al. examined whether somatic symptoms are overreported in PSD compared with primary depression (
8). A factor analysis on the Montgomery-Åsberg Depression Rating Scale revealed two factors that are considered to represent somatic and psychological symptoms of depression (SR4,
online supplement). There were no significant differences in these factors between patients with PSD and individuals with primary depression or between stroke patients and control participants without depression. The authors concluded that somatic symptoms should be considered in the diagnosis of PSD and that PSD is as valid a syndrome as primary depression (
8).
In contrast, Gainotti et al. compared the phenomenology of depression in stroke and primary depression and found more severe anxiety and greater frequencies of catastrophic reaction, hyperemotionalism, and diurnal mood variation in PSD and more suicidal ideation and anhedonia in primary depression (
9). These authors concluded that PSD includes more “motivated” symptoms (e.g., apathy), whereas primary depression includes more “unmotivated” symptoms (e.g., anhedonia). This proposal has several important limitations. First, the division of depressive symptoms into motivated and unmotivated categories is conceptually unclear and empirically unsubstantiated. Second, these authors used an ad hoc scale that included the symptoms of “catastrophic reaction” and “emotionalism,” which are not included in primary depression. Thus, the finding of higher frequencies of these symptoms in PSD compared with primary depression is a creation of the rating instrument. Third, anxiety and apathy are considered specifiers of major depression in DSM-5 but are not included as symptoms (
2). Thus, the finding of a different syndromic profile for PSD compared with primary depression is a result of an arbitrary nosological modification. Finally, the study by Gainotti et al. was flawed by making phenomenological considerations in the absence of statistical analyses (
9). The same research group proposed that the diagnosis of PSD relies upon the subjective report of specific symptoms, the external observation of the patient’s behavior, and the information supplied by depression inventories and scales (
10). This proposal is conceptually unclear: first, symptoms considered in isolation may lack adequate diagnostic specificity and sensitivity; second, the unspecified technique of “external” observation of putative “inner” feelings not only falls into Cartesian dualism but also has no empirical validation; and third, the validity of diagnosing depression with rating scale severity scores depends on the psychometric attributes of each instrument. Whereas Quaranta et al. criticized the use of instruments validated in primary depression for use in stroke (
10), most phenomenological studies in PSD have failed to find differences between primary depression and PSD. Furthermore, the use of generic psychiatric instruments and standardized diagnostic criteria for the diagnosis of PSD has relevant external validation, such as a significant improvement of PSD following treatment with antidepressants (
10,
11). Finally, the diagnostic accuracy of the Post-Stroke Depression Rating Scale used in the Quaranta et al. study was assessed against the Hamilton Depression Rating Scale (HAM-D), an instrument designed to rate the severity of primary depression (
10).
In DSM-5, PSD is listed as a depressive disorder caused by another medical condition, which is defined as a “prominent and persistent period of depressed mood or markedly diminished interest and pleasure in all, or almost all, activities that predominates in the clinical picture,” with the depressive disorder being “the direct pathophysiological consequence of another medical condition” (
2). Robinson and his research group have diagnosed PSD by using DSM-III and DSM-IV criteria for major and minor depression (
11). In DSM-5, the criteria for major depression were essentially unchanged, whereas minor depression was replaced by a “depressive episode with insufficient symptoms,” including depressed affect and at least one of the eight symptoms for a major depressive episode, that has a significant clinical impact and lasts for at least 2 weeks in the absence of a personal history of affective disorders. A mild type of depression has been frequently reported among stroke patients. Variously labeled minor, subsyndromal, or subthreshold depression, this entity is at the lower end of a spectrum of depression and depends on the quantity, quality, severity, and duration of depressive symptoms (
12). “Dysphoria” is another construct used in stroke and is conceptualized as ≥2 weeks of sad mood with loss of interest and anhedonia. The DSM-III and DSM-IV categories of minor depression include sad mood, anhedonia, or dysphoria and two additional symptoms of depression (
13).
In conclusion, our analysis suggests that PSD should be diagnosed by using the inclusive strategy and the DSM-5 criteria for major depression, whereas the DSM-5 criteria for depressive disorder with insufficient symptoms require validation.
Frequency of PSD
Before we examine the frequency of PSD, relevant diagnostic confounds, such as lesion type, age, aphasia, and recruitment characteristics, need to be discussed. Stroke may occur after thrombosis or thromboembolism of proximal arteries resulting in large strokes in the middle, anterior, or posterior cerebral arteries, which subsequently affect distal arterioles and result in lacunar strokes or ischemic lesions in watershed regions. Some strokes may present as sudden events with defined clinical manifestations or as covert or silent events with subtle or no clinical expression. Major depression, as defined by DSM-5, is frequently identified after strokes caused by large vessel occlusion and after silent lacunar infarctions (
14). Covert or silent strokes include small ischemic lesions or microbleeds that do not produce sensorimotor deficits (
14). Given that silent strokes occur five times more frequently than overt strokes, vascular depression (related to silent strokes) may occur more frequently than PSD (related to overt strokes) (
14). Moreover, the incidence of silent strokes among elderly depressed individuals is as high as 46% (
14). The type of stroke may also influence the phenomenology of depression because vascular depression is associated with more severe cognitive deficits, psychomotor retardation, apathy, guilt, loss of insight, and anhedonia than stroke without depression (
13).
Several studies have examined whether the phenomenology of depression is age related. Alexopoulos et al. found that older individuals may report more somatic and cognitive symptoms of depression than emotional symptoms (
15). Blazer and Williams reported a higher frequency of sad mood, loss of concentration, and psychomotor retardation, as well as decreased awareness of health problems, among older individuals with depression than among young individuals, a syndrome that, in the investigators’ opinion, did not fit any DSM category (
16). Finally, Amore et al. reported that elderly individuals with depression showed a high frequency of anxiety, nervousness, irritability, and importuning behavior (
13). Given that stroke is most frequent among elderly individuals, it is possible that the phenomenology of PSD is influenced by age, but it remains unclear whether diagnostic criteria should be modified.
Aphasia occurs in about one-third of stroke patients, which imposes a major challenge to the diagnosis of depression and thus prevents a more accurate estimation of prevalence. Several instruments have been adapted or designed to diagnose depression among individuals with aphasia, but most of these instruments have poor internal consistency, poor reliability, and unclear validity. The Stroke Aphasic Depression Questionnaire–Hospital Version (SADQ-H) is a 21-item instrument that was found to be valid and reliable in the diagnosis of depression among individuals with aphasia (SR5,
online supplement). One limitation of this instrument is that its validity was calculated in comparison with the Hospital Anxiety and Depression Scale, an instrument that is not valid or reliable among patients with severe comprehension deficits or global aphasia. Cobley et al. selected 10 items of the SADQ-H and found this abridged instrument to be reliable for PSD diagnosis, but its validity was not examined (SR5,
online supplement). Ashaie et al. used the Center for Epidemiologic Studies Depression Scale (CES-D) to diagnose “clinical” and subthreshold depression in a series of 144 stroke patients with aphasia and found that most patients with depression had Broca’s aphasia (65%), followed by anomic aphasia (13%), Wernicke’s aphasia (11%), and conduction aphasia (11%). There were no significant differences in the severity of aphasia among patients with major depression, subthreshold depression, or no depression (
17). Major depression was diagnosed among 19% of patients in the study sample, and subthreshold depression was diagnosed among 22%. Given the need for a basic level of verbal comprehension to answer questions for a reliable diagnosis of depression, the validity of the diagnosis of depression among individuals with severe aphasia cannot be determined.
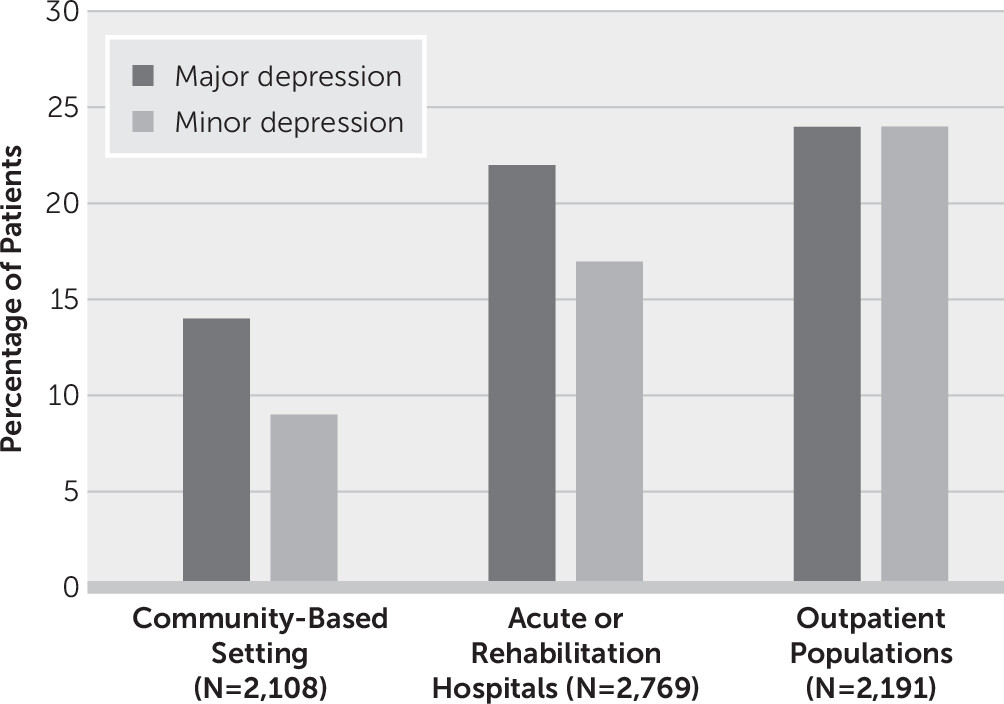
Several studies have examined the epidemiology of PSD. Robinson performed a pooled analysis of studies published prior to 2003 and included only studies that used structured interviews and diagnostic criteria to identify major and minor depression in community-based settings, acute or rehabilitation hospitals, or outpatient clinics (
18) (
Figure 1). The analysis demonstrated that the frequency of PSD is influenced by clinical setting and stroke severity. The frequencies for major and minor depression were 14% and 9%, respectively; in acute or rehabilitation hospitals, the frequencies were 22% and 17%; and in outpatient clinics, the frequencies were 24% and 24%.
A systematic review and meta-analysis that included data from 51 studies and 25,488 stroke patients provided a pooled frequency estimate for depression of 31% (95% CI=28–35) at any time point up to 5 years after stroke (
19). In this study, PSD was variously defined as depressive disorder, depressive symptoms, or psychological distress. On the basis of DSM-5 classifications, the disorders of major depression, minor depression, or dysthymia were diagnosed by using structured or semistructured psychiatric interviews (studies without categorical diagnoses were excluded). There was marked heterogeneity in the cutoff points for particular depression rating scales: for example, four different cutoff points were used with the Geriatric Depression Scale, the Beck Depression Inventory, and the Hospital Anxiety and Depression Scale, and three different cutoff points were used with the HAM-D (SR6–SR9,
online supplement). Similar pooled estimates of depression were reported for people with a history of depression before stroke and for individuals with aphasia.
Another analysis by Ayerbe et al. included 20,293 patients within 15 years after a stroke (
20). The study found a pooled prevalence of PSD of 29% (95% CI=25–32) at any time point; the prevalence of PSD was 22% in population studies, 30% in hospital studies and 30% in rehabilitation settings. The 5-year cumulative incidence of PSD was 42%, with a pooled prevalence of 29% that remained stable in the first 10 years after stroke (
20). The authors suggested that the source of variability in estimates of PSD prevalence mostly related to different diagnostic methods, source of patient recruitment, and time of assessment since stroke.
Hornsten et al. carried out a postal inquiry for people ≥65 years old in northern Sweden and Finland (SR10,
online supplement). The prevalence of stroke was 7%, and the prevalence of depression was 13%. The prevalence of PSD ranged from 11% among 65-year-old individuals to 18% among 80-year-old individuals, demonstrating that PSD increases with age. The prevalence ratio of depression in stroke was 1.77, but the estimate varied greatly depending on the definition of depression. Mitchell et al. carried out a meta-analysis on the frequency of PSD on the basis of ICD or DSM criteria and reported an estimate for major depression of 18%, minor depression of 13%, dysthymia of 3%, adjustment disorder of 7%, and any depressive disorder of 33% (
21). The frequency of PSD ranged from 16% in the community to 20% in rehabilitation settings, with a point prevalence of 18%. Cassidy et al. estimated the frequency of PSD by using the CES-D, the HAM-D, and a DSM-IV checklist (SR11,
online supplement). The frequency of major depression was 20% among patients undergoing inpatient rehabilitation, with a point prevalence of 3% in the general population (SR11,
online supplement). Taylor-Rowan et al. carried out a systematic review and meta-analysis on the prevalence of prestroke depression and PSD (
22). Prestroke depression was diagnosed by using the Structured Clinical Interview for DSM-IV, a specific cutoff score on valid depression scales, self-report, or methods of unclear validity (
22). A potential recall bias on the basis of the presence of PSD was not explored. The pooled prevalence of prestroke depression was 12%, which was associated with increased odds of PSD (odds ratio=3.0, 95% CI=2.3–4.0) (
22). Finally, Wu et al. examined the frequency of depression (as determined with the nine-item Patient Health Questionnaire) among 243 patients with silent lacunar infarction and found a frequency of PSD of 25% (SR12 and SR13,
online supplement).
In conclusion, the epidemiology of PSD depends on diagnostic instruments and use of diagnostic criteria, and it is influenced by the presence of aphasia, age, and type of stroke. Regardless of these confounders, most epidemiological studies and meta-analyses suggest a prevalence of PSD of approximately 30%.
Effect of PSD on Stroke Recovery and Mortality
Numerous studies have examined the relationship between depression at initial examination and functional and motor outcomes (
44) (SR20 and SR21,
online supplement). Five of six studies that examined the relationship between depression at baseline and ADLs at 1 or more years of follow-up found that depression severity was an independent predictor of severity of ADL impairment.
Consistent with these findings, patients with PSD who responded to treatment with nortriptyline or fluoxetine showed significantly better improvement in ADLs than patients with PSD given placebo (
45). Furthermore, response to treatment of PSD with nortriptyline or fluoxetine was associated with greater cognitive recovery over 2 years compared with changes associated with placebo administration (SR22,
online supplement).
PSD also leads to increased mortality. The first study to report this phenomenon was published in 1993 (
46). In this study, 103 patients were followed up at 10 years after their index stroke to determine mortality rates. Patients who developed PSD immediately after stroke had higher mortality rates than equally impaired stroke patients with no in-hospital depression (odds ratio=3.4, 95% CI=1.4–8.4, p=0.007) (
46). A similar finding was reported by House et al. who showed that even mild severity of PSD was associated with increased mortality as early as 1 year after stroke (SR23,
online supplement). Furthermore, in a cohort of 51,119 veterans with ischemic stroke, patients who developed PSD had a higher 3-year mortality risk than those without a mental health diagnosis (
47).
A recent study of 1,354 patients that used the Hospital Anxiety and Depression Scale (HADS; higher scores indicate greater impairment) to assess PSD found increased mortality at the 5-year follow-up among patients with HADS scores ≥7 at 3 months after stroke compared with patients with scores <7 at 3 months (hazard ratio=1.41, 95% CI=1.13–1.77, p=0.02) (
48) (SR24,
online supplement). The investigators also reported increased mortality among patients who were started on selective serotonin reuptake inhibitors (SSRIs) in the 3 months after stroke. However, this was not a randomized trial, and the data analysis on which this assertion was based did not control for all the relevant confounders, such as level of disability, depression severity, and the effects of comorbid medical conditions (
48).
The association of PSD with mortality appears to be the result of an increase in cardiovascular mortality (
48). We reported that decreased heart rate variability, which has been demonstrated to play an etiological role in mortality associated with depression and myocardial infarction, was also associated with PSD (
49,
50). Thus, disruption of autonomic system function among patients with PSD may contribute to cardiovascular mortality.
Antidepressant use may influence long-term survival after stroke (
51). A 9-year follow-up study of patients with or without PSD who had been treated with nortriptyline (100 mg/day) or fluoxetine (40 mg/day) early after an index stroke showed that patients receiving active treatment (N=53) had an increased probability of survival at the 9-year follow-up compared with similar patients given placebo (N=28) (i.e., a 59% 9-year survival rate among patients treated with nortriptyline or fluoxetine versus a 35% rate among patients given a placebo; adjusted odds ratio=3.7, 95% CI=1.1–12.2, p=0.03) (
51). A beneficial effect of antidepressants on long-term survival after stroke was also observed in a group of 790 veterans with stroke who were followed over a 7-year period (SR25,
online supplement).
Effect of Antidepressants on Stroke Recovery
Many basic and animal studies have shown that SSRIs have consistent effects on neural plasticity, including neurogenesis, synaptic remodeling, and network reorganization (
52) (SR26 and SR27,
online supplement). These effects are mediated, at least in part, by the activation of neurotrophic-signaling pathways (e.g., BDNF, tropomyosin receptor kinase B receptors, and translation factors such as cAMP response element-binding protein), as well as reductions in oxidative stress and neuroinflammation (
53). Furthermore, SSRIs and other small molecules were shown to induce, in adults, a form of plasticity that is typically observed during developmental critical periods (
54).
A recent systematic review and meta-analysis of data from studies examining the effects of antidepressants in animal models of stroke (44 publications and 22 medications) found that antidepressants reduced infarct volume by 27% (95% CI=21–34) and enhanced motor and behavioral outcomes by 53% (95% CI=46–61). SSRIs, the most frequently studied antidepressants, improved neurobehavioral outcomes by 52% (95% CI=39–65) (
55) (SR28 and SR29,
online supplement).
In humans, antidepressants have been reported to improve motor, cognitive, and functional outcomes. For example, the Fluoxetine for Motor Recovery After Acute Ischemic Stroke trial examined the efficacy of fluoxetine to enhance motor recovery following ischemic stroke (
56). In this double-blind, placebo-controlled trial, 118 stroke patients undergoing physical rehabilitation were randomly assigned to receive fluoxetine (20 mg/day) or placebo for 3 months starting 5–10 days after the onset of stroke. The primary outcome measure was the change in Fugl-Meyer Motor Scale scores (FMMS; lower scores indicate more severe impairment) following the intervention. The improvement in FMMS score at day 90 was significantly greater in the fluoxetine group than in the placebo group. Furthermore, the number of patients that achieved independence (defined as a score of 0–2 on the modified Rankin Scale [mRS]) was higher in the fluoxetine group (
56).
Our group examined the efficacy of escitalopram to improve cognitive outcomes following stroke. The 12-month trial included 129 stroke patients without depression who were randomly assigned to three arms: a double-blind, placebo-controlled comparison of escitalopram (N=43) with placebo (N=45) and a nonblinded arm engaged in problem-solving therapy, a form of psychotherapy (N=41). The primary outcome measure was change in score on the Repeatable Battery for the Assessment of Neuropsychological Status (RBANS; with lower scores indicating greater impairment) (
57).
When compared with patients who received placebo or underwent problem-solving therapy, stroke patients who received escitalopram improved in global cognitive functioning, immediate memory, and delayed memory (
57). These beneficial effects of escitalopram were independent of escitalopram’s effect on depression. Furthermore, there was a positive, but not statistically significant, correlation between change in RBANS total score and change in Functional Independence Measure total score (Spearman’s correlation=0.25, p=0.20). Thus, the reported changes in neuropsychological test scores were associated with a measurable, albeit not statistically significant, improvement in ADLs.
A recent meta-analysis of 52 trials examined the efficacy of SSRIs to reduce disability among 4,060 participants with a recent stroke. The authors concluded that SSRIs may improve recovery after stroke. However, larger rigorous trials are needed to replicate these findings, as well as to determine the safety of SSRIs in the context of stroke (
58).
The cumulative evidence suggesting a beneficial effect of fluoxetine on stroke recovery led to three large multicenter randomized controlled trials (RCTs) in the United Kingdom (Fluoxetine or Control Under Supervision [FOCUS]), Sweden (Efficacy of Fluoxetine–a Randomised Controlled Trial in Stroke [EFFECTS]), and Australasia (Assessment of Fluoxetine in Stroke Recovery [AFFINITY]) (SR30, online supplement). These trials share essential methodological aspects and provide individualized and combined results on the efficacy of fluoxetine to reduce dependency and disability among patients with recent stroke. The targeted enrollment across the three studies was 6,000 participants. The actual enrollment amounted to 4,344 participants.
In the FOCUS trial, 1,564 patients with a recent stroke and focal neurological deficits were allocated to the fluoxetine (20 mg/day) group, and 1,563 patients were allocated to a matching placebo group (
59). The primary outcome was functional status, measured with the mRS, at 6 months. Fluoxetine was not better than placebo in reducing disability following stroke, although it effectively prevented depressive disorders (
59).
The AFFINITY trial was conducted in hospitals in Australia, New Zealand, and Vietnam. A total of 1,280 patients with recent stroke (ischemic or hemorrhagic) were randomly assigned to fluoxetine (20 mg/day) or placebo groups for 6 months. There were no significant differences in disability outcomes measured with the mRS. On the other hand, the patients receiving fluoxetine had a higher risk of falls, bone fractures, and epileptic seizures than patients receiving placebo (
60) (SR31,
online supplement).
The EFFECTS trial was conducted in 35 stroke and rehabilitation centers in Sweden. A total of 1,500 patients were allocated to receive fluoxetine (20 mg/day) or placebo and followed for 6 months. Although fluoxetine was efficacious in preventing depression, there were no significant effects on disability outcomes. In addition, patients receiving fluoxetine had a higher frequency of bone fractures and hyponatremia (
61).
The findings of these trials provide consistent evidence that fluoxetine is not better than placebo in improving disability outcomes measured with the mRS. However, we need to consider certain limitations that temper the clinical implications of these findings. First, the three trials enrolled patients with milder forms of stroke (the median National Institutes of Health Stroke Scale scores were 3, 6, and 6 for the EFFECTS, AFFINITY, and FOCUS trials, respectively), making them more vulnerable to ceiling effects. Second, although the mRS is an established measure of stroke-related disability, the scale is not sensitive to the occurrence of more subtle changes (e.g., cognitive, motivational, and autonomic changes) that may have an effect on long-term outcomes. Finally, it is important to note that these pragmatic trials did not require participants to be involved in rehabilitation therapy, nor was the intensity of rehabilitation procedures recorded. This is a drawback because the mechanisms of action of SSRIs in stroke recovery involve activity-dependent plasticity, and thus, the network reorganization associated with fluoxetine depends on the concurrent performance of rehabilitation practices.
Etiological Mechanisms of PSD
There is consensus that psychological, social, and biological factors contribute to the mechanisms underlying PSD (SR32,
online supplement). The occurrence of PSD has been consistently associated with stroke severity and the degree of functional physical and cognitive impairment (
26).
Biological factors contributing to PSD include alterations in ascending monoaminergic systems, hypothalamic-pituitary-adrenal (HPA) axis abnormalities, disruption of prefrontal-subcortical circuits, alterations in neuroplasticity and glutamate neurotransmission, and excessive levels of proinflammatory cytokines (
62–
64) (SR33–SR35,
online supplement). However, a pathophysiological hypothesis of PSD that can integrate these changes into a coherent explanatory model has yet to be formulated.
From a translational approach, there have been several attempts to model PSD in experiments with rodents. We conducted the earliest studies using a rat model of middle cerebral artery (MCA) ligation. Right MCA ligation produced significant bilateral depletion of norepinephrine and dopamine in both the cortex and brainstem, as well as hyperactivity and other behavioral changes not seen in rats given sham ligations (
65). Subsequent experiments using this animal model demonstrated that the effects on biogenic amines and behavior were lateralized—they did not occur after left MCA ligation—and lasted up to 4 weeks (SR36,
online supplement).
In recent years, there has been renewed interest in modeling the neuropsychiatric consequences of stroke. Kronenberg et al. used a mouse model of mild stroke (i.e., a 30-minute MCA occlusion that limits the ischemic damage to the basal ganglia) (
66). A subgroup of these mice was given citalopram, starting 1 week after MCA occlusion. Left, but not right, MCA occlusion produced delayed degeneration of dopaminergic neurons in the left ventral tegmental area, which resulted in reduced dopamine concentrations in the striatum. The behavioral correlates of these neurodegenerative and neurochemical changes were anhedonia and behavioral despair assessed with the sucrose consumption test and forced swim test, respectively. Importantly, chronic citalopram treatment initiated 7 days after stroke prevented the degeneration of dopaminergic neurons and reversed the behavioral phenotype (
66).
There have also been animal studies that combined experimental ischemic lesions with social isolation or chronic mild stress protocols implemented during the stroke recovery process (
67) (SR37,
online supplement). Permanent left MCA occlusion coupled with a 14-day chronic mild stress protocol produced a depressive phenotype characterized by decreased exploratory behavior and sucrose consumption These behavioral effects were reversed by the administration of citalopram and a serotonin-1A antagonist, and there was evidence of increased neurogenesis in the hippocampus (
68).
In a rodent model of PSD, electrical stimulation of the cerebellar fastigial nucleus decreased depression-like symptoms and increased regional cerebral blood flow (SR38, online supplement). Fastigial nucleus stimulation was associated with a reduction in the expression of inflammatory cytokines (SR39, online supplement).
Unfortunately, there have been relatively few studies on the mechanisms underlying PSD in clinical populations. However, one study of PSD conducted by our group showed decreased cerebral spinal fluid levels of serotonin or norepinephrine metabolites that were significantly associated with severity of PSD (
69).
There is a growing consensus that ischemic lesions (single or multiple) of the neural circuits that connect the prefrontal cortex, basal ganglia, thalamus, and amygdala (independent of their lateralization) may disrupt mood regulation and executive function, leading to similar clinical presentations. Furthermore, there appears to be a threshold by which the confluence of multiple etiological factors or further damage to specific white matter tracts, such as the cingulate bundle, the uncinate fasciculus, and the superior longitudinal fasciculus, triggers the onset of clinical depression. Thus, acute ischemia can reveal the presence of vascular depression (SR33, online supplement).
From a network standpoint, resting connectivity patterns revealed by functional MRI appear to be similar in vascular depression and PSD, with increased activation of the default mode network and limbic structures and decreased activation of task-related networks and the dorsolateral aspects of the prefrontal cortex (
70).
Earlier studies also examined dysregulation of the HPA axis in PSD, particularly the association of PSD with elevated cortisol levels and abnormal negative feedback control of cortisol secretion (
63). More recently, several studies emphasized the role of proinflammatory cytokines in the development of PSD (
62) (SR35 and SR40–SR44,
online supplement). Increased serum concentrations of interleukin-6 were associated with increased severity of somatic symptoms of PSD, and increased levels of inflammatory cytokines reduce the synthesis and availability of serotonin through their enhancing effect on the activity of the enzyme indolamine 2,3-dioxygenase. A high neutrophil-to-lymphocyte ratio on admission, measured systemically, predicted PSD after 1 month and 6 months (SR45 and SR46,
online supplement).
In summary, although there are numerous possible physiological mechanisms related to PSD, many investigators have concluded that this complex disorder, like most major psychiatric disorders not associated with stroke, may best be described as a biopsychosocial disorder. However, this general theoretical framework does little to help us elucidate pathophysiological mechanisms leading to specific symptoms. The different etiological factors described above may have more salient roles in some forms or symptoms of PSD, and their effects may vary at different times after stroke. We believe future studies should attempt to identify the mechanisms underlying specific symptoms or clinical characteristics of PSD rather than the whole syndrome.
Treatment of PSD
Selected RCTs of PSD and vascular depression are shown in
Table 1 (
33,
71–
83). The first randomized double-blind treatment trial was reported in 1984 by Lipsey et al. (
71). Patients randomly assigned to the nortriptyline group (50–100 mg/day) had a significantly greater reduction in HAM-D scores (indicating lower severity) over 6 weeks of treatment compared with patients in the placebo group. The first double-blind controlled trial to examine the efficacy of SSRIs was reported by Andersen et al. in 1994 (
73). Among 33 poststroke patients given citalopram (10–20 mg/day), there was a significantly greater reduction in HAM-D scores over 6 weeks among 33 poststroke patients given citalopram (10–20 mg/day) than among 33 patients with similar levels of depression given placebo.
A recent meta-analysis of 16 RCTs (with 12 trials using antidepressants and four evaluating the efficacy of psychotherapy) that included 1,655 patients found a significant beneficial effect of antidepressant medication, whereas psychotherapy was not more effective than a control intervention (
84). Brief psychosocial therapies, however, that place an emphasis on care management, psychoeducation, and family support can be beneficial when administered in combination with antidepressants to treat or prevent PSD (SR47,
online supplement).
Recent small RCTs of escitalopram and citalopram also showed beneficial effects on depressive symptoms and endothelial function (
85) (SR48,
online supplement). In addition, a clinical trial examining the efficacy of intravenous alteplase on PSD showed a significant reduction in the severity of depressive symptoms 3 months after an index stroke (SR49,
online supplement). Similarly, pioglitazone (a peroxisome proliferator-activated receptor-gamma agonist) decreased the severity of PSD symptoms among stroke patients with diabetes mellitus type 2 (SR50,
online supplement).
It should be acknowledged, however, that treatment with antidepressants is not without risk. For example, SSRI use has been associated with an increased risk of hemorrhagic complications and an increased risk of falls among the elderly population (
86). Furthermore, other epidemiological studies have reported that SSRIs are associated with increased risk of stroke, myocardial infarction, and all-cause mortality (
87) (SR51,
online supplement). The effects of these antidepressants may be due to interactions with other variables, such as depression, disability, and comorbid medical conditions, that require further elucidation.
There have been recent small studies of nonpharmacological interventions to treat PSD. Nguyen et al. used cognitive-behavioral therapy and reported a significant decrease in depressive symptoms, along with improvements in fatigue and sleep quality (
88). Environmental enrichment and gamified training with virtual reality and other advanced technological tools demonstrated beneficial effects for patients with PSD (
89) (SR52,
online supplement). Behavioral activation therapy, which aims to prolong the frequency of pleasant or enjoyable events, also showed some promising results (SR53,
online supplement). Preliminary data from group-based acceptance and commitment therapy and self-help relaxation seem to confer certain positive benefits in terms of PSD resolution (
90) (SR54,
online supplement). A social support and health education program using a two-pronged approach of providing rehabilitation-related information support and emotional support appears to help when provided over a minimum of 8 weeks of therapy (SR55,
online supplement). Reminiscence therapy—aimed at recalling important life events to improve pleasure and belonging—also appears to help in reducing the PSD burden (SR56,
online supplement). We have previously shown that repetitive transcranial magnetic stimulation of the left dorsolateral prefrontal cortex may be efficacious in treating PSD (
91). These findings were replicated in a recent independent study (
83). In addition, transcranial direct current stimulation of the dorsolateral prefrontal cortex was shown to be safe and efficacious in reducing the severity of PSD (
92). Studies of transcranial alternate current stimulation are also under way (SR57,
online supplement). Finally, both aerobic exercise and aquatic therapy have been employed to decrease the burden of PSD (
93) (SR58 and SR59,
online supplement).