Definition of typical and atypical antipsychotic drugs
Traditional or “typical” antipsychotics such as haloperidol and chlorpromazine, when used in clinically effective dosages, induce elevated levels of serum prolactin, extrapyramidal signs and symptoms (EPS) and, after a period of time, tardive dyskinesia (TD). However, antipsychotic drugs such as olanzapine, clozapine, quetiapine, and amisulpride are “atypical” because, in contrast to the traditional antipsychotics, they elicit low or negligible levels of these untoward side effects while still effectively controlling psychotic symptoms.
Which neuron pathway is clinically most affected by antipsychotic drugs?
Immediately after the clinical introduction of drugs for psychosis (
1), clinicians observed that patients taking these medications exhibited a Parkinson-like syndrome of tremor, akinesia, and rigidity (
2). This drug-induced parkinsonism strongly suggested that antipsychotic drugs were interfering with dopamine pathways in the human brain, because Parkinson’s disease was known to be a disease of insufficient dopamine neurotransmission. This clinical observation gave birth to the dopamine hypothesis of psychosis and antipsychotic drug action (
3).
Although it was suggested that chlorpromazine and haloperidol blocked “5-hydroxytryptamine (serotonin) and monoaminergic (noradrenaline and dopamine) receptors” (
4), it was not possible at that time to conclude which of the 3 pathways was selectively affected by antipsychotics. This is because the turnover of noradrenaline, serotonin (5-HT), and dopamine were all simultaneously affected by the antipsychotics (
4,
5). Andén and others speculated that chlorpromazine and haloperidol “reduce the elimination rates of these” metabolites of noradrenaline, 5-HT, and dopamine (
5). Although Andén and others (
6) subsequently found that antipsychotic drugs in vivo had a greater effect on dopamine turnover than on noradrenaline turnover, direct in vitro evidence for the selective blockade of dopamine receptors was found only later (
7–
9).
The multiple clinical and adverse effects of various antipsychotic drugs depend on the combination of receptors occupied, but the dopamine pathway is the primary common target for all antipsychotic drugs. More specifically, “no drug has yet been identified with antipsychotic action without a significant affinity for the D
2 receptor” (
10,
11).
Which group of dopamine receptors is clinically relevant for antipsychotic drug action?
There are 5 types of dopamine receptors in human beings (
12,
13). Types 1 and 5 are similar in structure and drug sensitivity (
14,
15), and these 2 receptors are referred to as the “D
1-like” group or class of receptors. Dopamine receptor types 2, 3, and 4 are also similar in structure and are, therefore, grouped together as the “D
2-like” group. Dopamine receptors 2, 3 and 4, however, have significantly different sensitivities to antipsychotic drugs.
Al though the D
1-like receptors are often mentioned as a primary target for antipsychotic drugs (
16), 3 findings indicate that the D
1-like receptors are not clinically relevant in the therapeutic action of these drugs. First, D
1 antagonists do not clinically improve psychotic signs and symptoms (
17–
19). Second, therapeutic maintenance dosages of various antipsychotic drugs occupy low or negligible levels of D
1 receptors in the brains of patients with psychosis (
20). For example, therapeutic dosages of haloperidol occupy less than 5% of the dopamine receptors in the brain putamen of schizophrenia patients (
20). Although therapeutic dosages of some antipsychotic drugs, such as clozapine, occupy approximately 36% to 59% of brain dopamine D
1 receptors (
21), there is no currently known reason to believe that these occupied D
1 receptors contribute to the unique properties of clozapine. Third, for the D
1 dopamine receptor, the binding constants (that is, the dissociation constants, also referred to as the inhibition constants, or Ki values) of various antipsychotic drugs (
22) are very much higher than the concentrations of antipsychotic drugs found in the cerebrospinal fluid or in the plasma water of patients (
12,
23,
24). In other words, if the free concentrations of antipsychotic drugs were as high as the values for the binding constants at D
1, the drugs would be toxic or lethal to patients.
Of the 3 D
2-like receptors, only the D
2 receptor itself is blocked by antipsychotic drugs in direct relation to their clinical antipsychotic potencies (
8,
9,
25). Although this long-known relation is sometimes criticized as simply a relation between the D
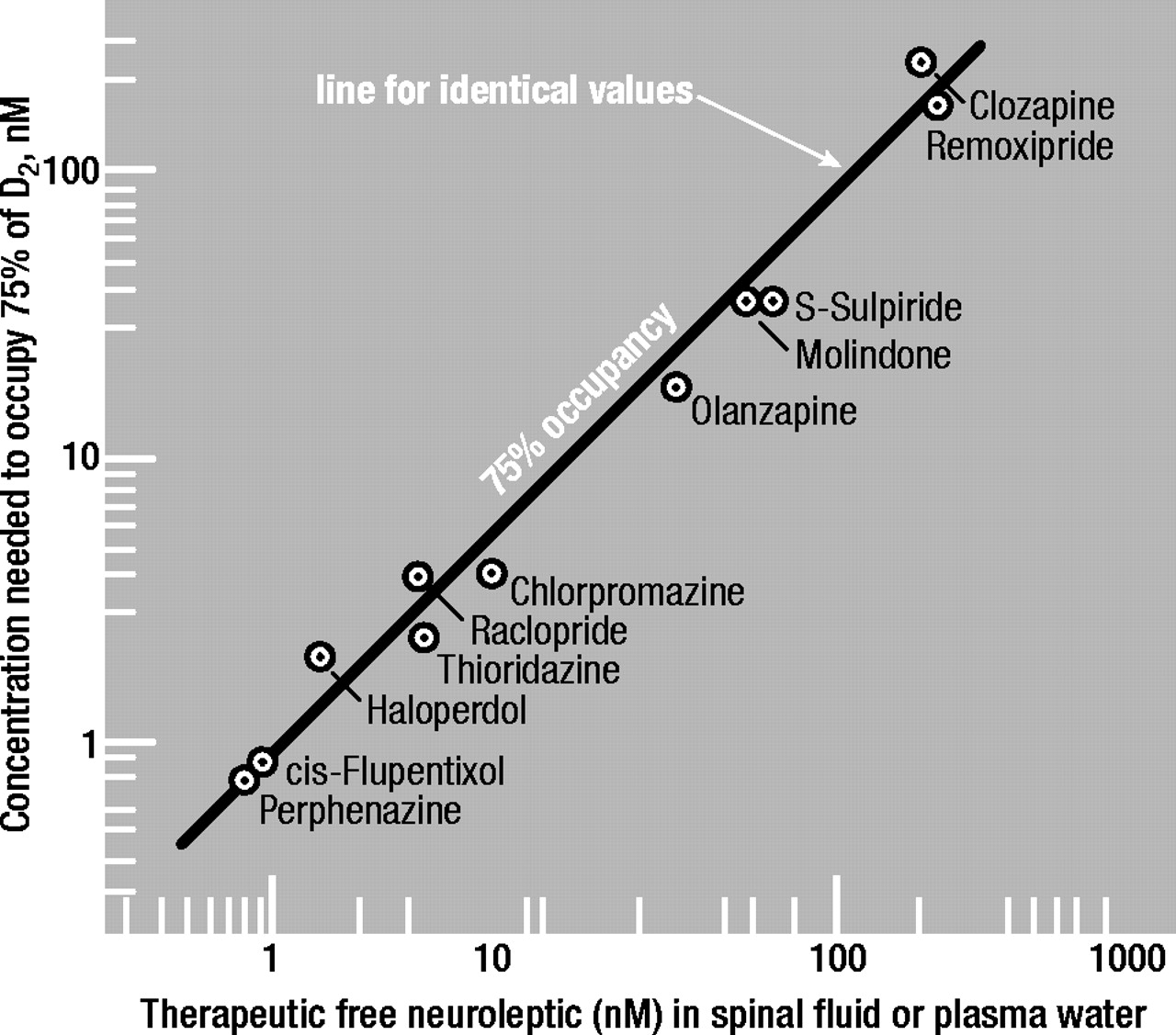
2-blocking concentrations and the clinical dosages at which EPS first appear, it is important to note that the concentrations of antipsychotics which block D
2 receptors in the brain are precisely identical to the concentrations found in the spinal fluid or plasma water (that is, corrected for drug binding to the plasma proteins) of patients whose psychotic symptoms are successfully controlled by antipsychotics (Figure 1). Because it is known that the clinical efficacy of antipsychotics is associated with a blockade of 60% to 80% of D
2 receptors in the brain (
26–
28), the antipsychotic concentrations shown on the ordinate in Figure 1 are those needed to block 75% of D
2 receptors in the presence of a physiological concentration of dopamine (see next section).
Endogenous dopamine raises the antipsychotic concentration needed for D2 block
Upon entry into the synaptic space, the antipsychotic drug must compete with endogenous dopamine for the receptor. Thus, the antipsychotic therapeutic concentration needed to block 50% of dopamine receptors in the presence of dopamine will be higher than that needed in the absence of dopamine. This is in accordance with the equation C50% = Ki ′ [1+D/D
2high], where D is the dopamine concentration in the synaptic space and where D
2high is the dissociation constant of dopamine at the high-affinity state of the dopamine D
2 receptor. The level of dopamine in the synaptic space in humans is not known but, in the rat nucleus accumbens, it is between 1 and 4 nM at rest, momentarily rising to 200 nM for the few milliseconds it takes for a nerve impulse to propagate (
29). The dopamine D
2 receptor can exist in either a high-affinity state or a low-affinity state for dopamine. The high-affinity state, D
2high, is the physiologically functional state (
30). The dissociation constant of dopamine at D
2high is 1.75 nM (see later).
Hence, although the concentration of dopamine, D, in the human synaptic space is not known, it appears that D is of the same order of magnitude as the dopamine Ki for D2high. Hence, with this single assumption that D is equivalent to D2high, the above equation of C50% = Ki ′ [1 + D / D2high] reduces to C50% is equivalent to 2 ′ Ki. The fraction, F, of D2 receptors occupied by an antipsychotic at a concentration C is C / (C + Ki). Using this formula, it can be shown that the concentration of an antipsychotic drug needed to occupy 75% of the D2 receptors is about 3 times higher than that required to occupy 50% of the receptors.
Therefore, using the above equations, the antipsychotic concentrations to occupy 75% of D2 receptors in patients were calculated and found to be virtually identical to the therapeutic concentrations of the antipsychotic drugs in the cerebrospinal fluid or in the plasma water (that is, corrected for drug binding to the plasma proteins) of patients being successfully maintained on these medications. This is illustrated in Figure 1.
“Fast-off” theory of atypical antipsychotic action: atypicals are rapidly released from D2 receptors
As noted above, clinically effective dosages of antipsychotic drugs occupy between 60% and 80% of brain dopamine D
2 receptors in patients, as measured by positron emission tomography (PET) or single photon emission tomography (SPET) in the human striatum (
28,
32–
37,
38–
53). Clozapine and quetiapine, however, have consistently been apparent exceptions. For example, in patients taking therapeutically effective antipsychotic dosages of clozapine, this drug only occupies between 0% and approximately 50% of brain dopamine D
2 receptors, as measured by various radioligands using either PET (
26,
28,
31,
34–
36,
54–
58) or SPET (
47–
52,
59).
Because the atypical antipsychotics occupy many different types of receptors under therapeutic conditions, any apparent exceptions to the “60% to 80%” rule of D
2 occupancy must be taken seriously. For example, the apparently low occupancy of D
2 by clozapine might suggest that D
2 is not the major antipsychotic target for clozapine (
31,
60). This is an important point because, if D
2 is not the common target for all antipsychotic drugs, then the explanation for atypicality must be found elsewhere, perhaps in the 5-HT system or in the balance between 5-HT and dopamine.
However, the apparently low occupancy of D
2 by clozapine and quetiapine is readily explained by the fact that these 2 antipsychotics rapidly dissociate from the dopamine D
2 receptor (
27). This also holds for remoxipride and amisulpride, 2 atypical drugs not used clinically in Canada. For example, human-cloned dopamine D
2 receptors release [
3H]clozapine, [
3H]quetiapine, [
3H]remoxipride, and [
3H]amisulpride at least 100 times faster than they release [
3H]haloperidol or [
3H]chlorpromazine (
27,
61,
62).
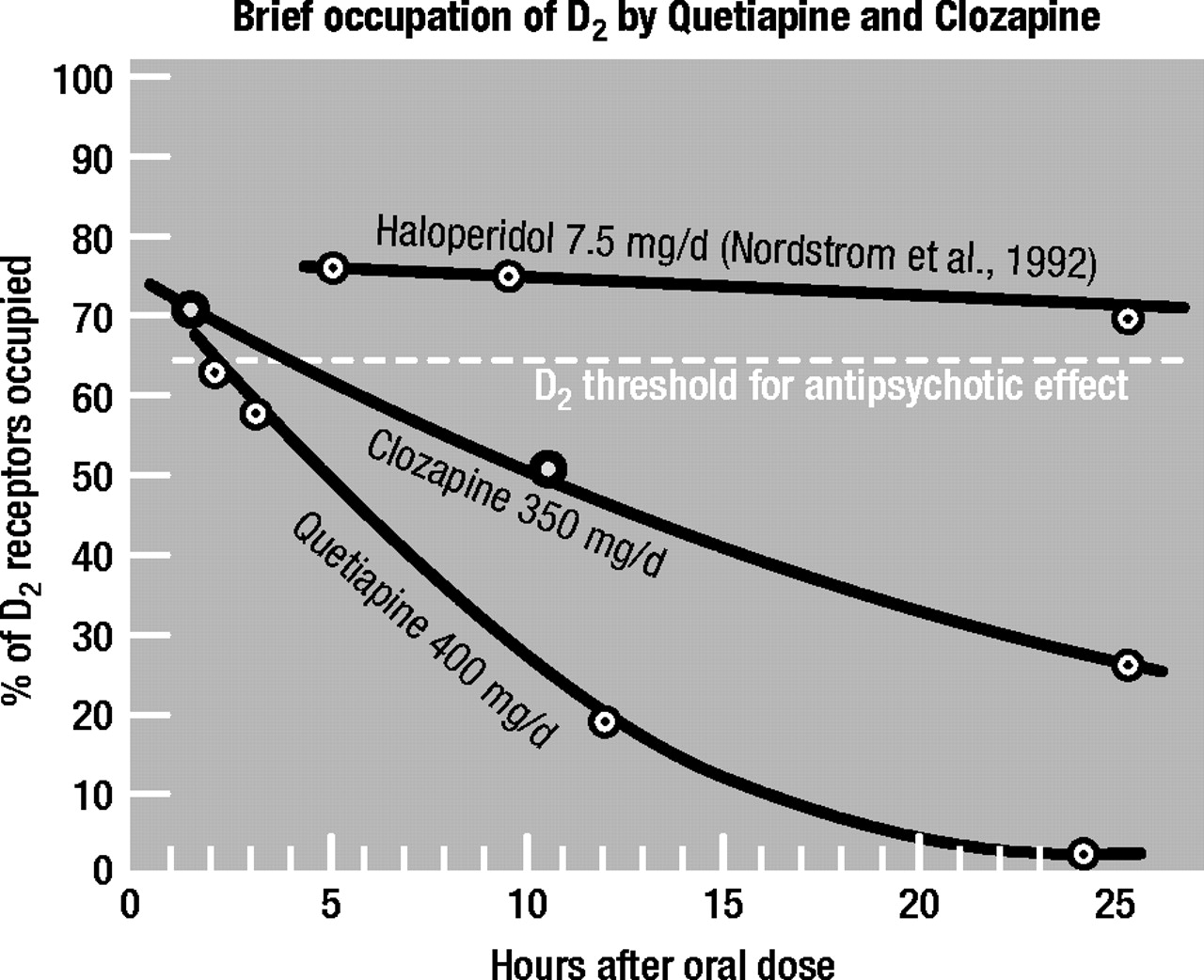
Figure 2 shows the rapid release of [3H]clozapine, [3H]quetiapine, [3H]remoxipride, and [3H]amisulpride from human-cloned dopamine D2 receptors; the slow release of [3H]raclopride, [3H]haloperidol, and[3H]chlorpromazine; and the intermediate release rates from these receptors for [3H]olanzapine and [3H]sertindole.
These in vitro data match those found clinically for clozapine, quetiapine, and haloperidol in schizophrenia patients and healthy volunteers. This is shown in Figure 3, where it has been found by PET (using [
11C]raclopride) that the human brain (striatum) occupancy of D
2 by quetiapine and clozapine rapidly falls off within 24 hours, in contrast to that for haloperidol, which maintains its D
2 occupancy constant over 24 hours (
38,
53,
63).
Thus, the rapid release of clozapine and quetiapine from dopamine D2 receptors and their replacement by endogenous dopamine would readily account for the low D2 receptor occupancy shown by these atypical antipsychotics.
It is important to emphasize that the rapid release of clozapine and quetiapine is a molecular event which occurs quickly, regardless of the clinical dosage used. In other words, even though high dosages of clozapine and quetiapine may be used, these drugs continue to go on and off the D2 receptor rapidly, allowing extensive and frequent access of endogenous dopamine to the receptor.
Hence, it appears that some antipsychotics, such as clozapine and quetiapine, occupy D2 receptors only transiently throughout the day. As just mentioned, PET imaging of patients with schizophrenia reveals that the D2 receptor occupancies by clozapine and quetiapine wear off quickly after an oral dosage, and patients may show no occupancy whatsoever within 48 hours of the last dose, in contrast to typical antipsychotics, which may continue to occupy D2 receptors for days. This may explain why psychotic relapses of patients on clozapine and quetiapine occur soon after withdrawal of the antipsychotic (64, 65, reviewed in 27)—much earlier than after withdrawal of conventional antipsychotic drugs such as haloperidol or chlorpromazine.
Clinical and basic implications of the “fast-off” theory of atypical antipsychotic action
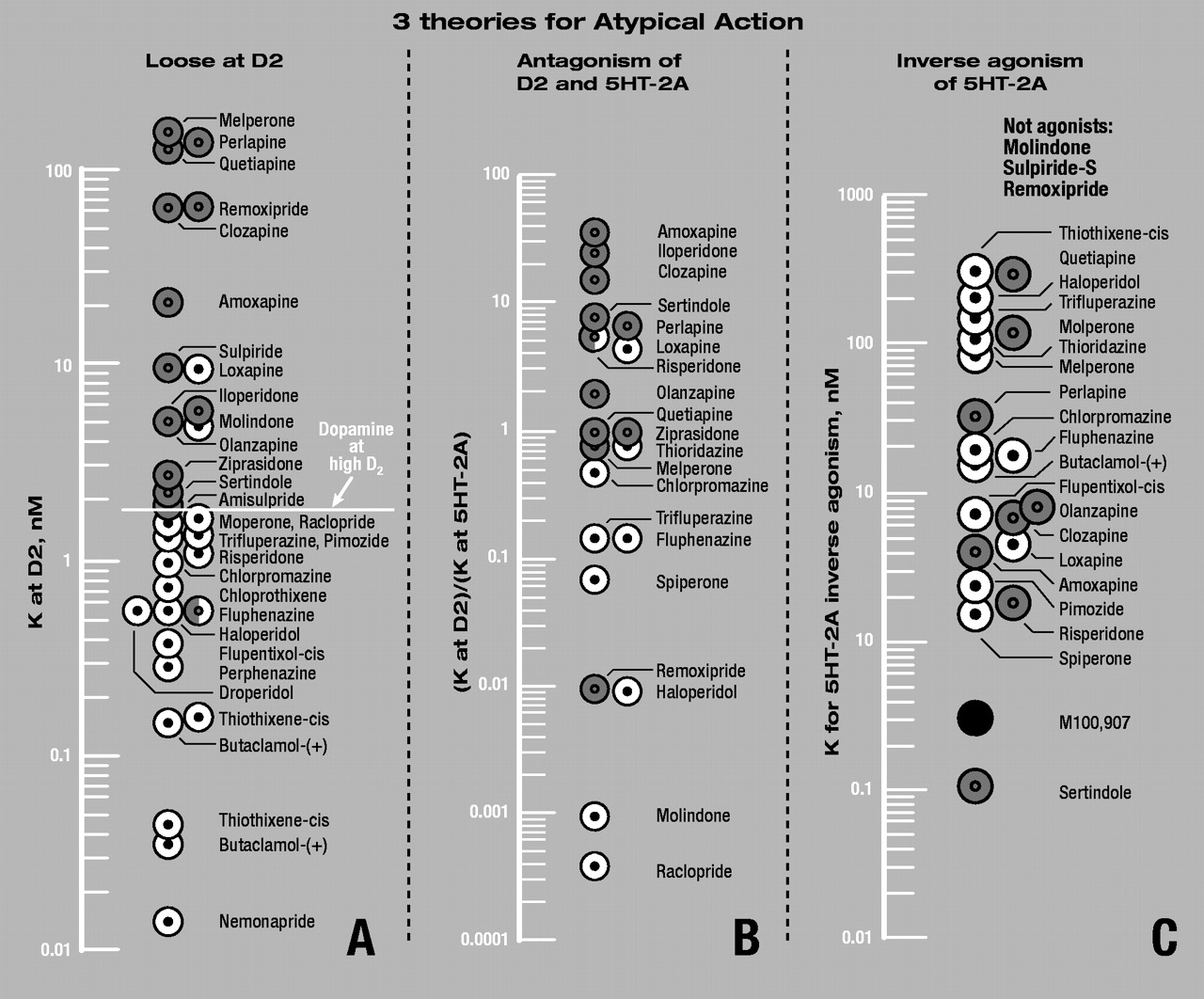
As outlined above, the “fast-off” theory of atypical antipsychotic action is that the atypicals have low affinities for the dopamine D2 receptor, and are loosely bound to, and rapidly released from, these receptors. A critical aspect of the theory is that the atypical antipsychotics bind more loosely to D2 than does dopamine it self, while the traditional, typical antipsychotics bind more tightly than dopamine. These data are summarized in Table 1 and Figure 4A.
Figure 4A illustrates a general demarcation between typicals and atypicals. That is, the typical antipsychotics have K values lower than that for dopamine (at the high-affinity state of the D
2 receptor), while the atypicals have K values higher than that for dopamine. Although risperidone appears to be an exception to this generalization, risperidone is the weakest “atypical” antipsychotic, eliciting dosage-dependent EPS in 60% to 70% of patients taking 6 mg or more daily, a dosage that may be insufficient for clinical efficacy (
66).
Clearly, the separation between typicals and atypicals in Figure 4A is not sharp and precise, because antipsychotic drugs with K values between 2 nM and 10 nM (including molindone and loxapine) often reveal dose-dependent EPS. Thus, the demarcation between typical and atypical antipsychotics is not a sharp divide but rather a continuous one. Antipsychotics become increasingly more atypical as their binding to the D2 receptor becomes more loose and they are released more quickly. Although all atypical antipsychotics have loose binding, with dissociation constants looser than 1.8 nM/L, they can still elicit dosage-dependent parkinsonism. For example, olanzapine, with a dissociation constant of 5.1 nM, is known to be associated with a dose-dependent incidence of EPS in some patients and especially at higher dosages. If the binding is extremely loose, as with clozapine, remoxipride, quetiapine, and melperone, essentially no EPS occurs (although exquisitely sensitive patients do exist who will exhibit EPS even with these drugs). Drugs that are too loose or have far too low an affinity for D2 receptors cease to exhibit any antipsychotic activity at all. Moreover, although the degree of occupancy of atypicals at D2 receptors has a direct influence on EPS, the potent anticholinergic action of olanzapine and clozapine provides an additional anti-EPS mechanism. It is because of its anticholinergic properties, for instance, that thioridazine use is relatively free of EPS.
Table 2 summarizes a few clinical distinctions between the typical antipsychotics, which are tightly bound to D
2, and the atypical antipsychotics, which are loosely bound to D
2. The required antipsychotic dosage (in mg) will be low for tightly bound drugs but high for loosely bound drugs. The typicals, being tightly bound to D
2, will elicit EPS and elevated prolactin, while the atypicals, being loosely bound and rapidly released from D
2, will not elicit these side effects, or will at least elicit them to a markedly lesser extent. Finally, because the typicals remain attached to D
2 and readily accumulate in brain tissue, they will eventually lead to TD (
67). The atypicals, however, are much less fat-soluble, and because they are readily released from D
2 and from the brain tissue, the risk of causing TD is much reduced or perhaps absent.
l-Dopa psychosis: “fast-off-D2” theory predicts low dosage of atypical antipsychotics
The treatment of patients with psychosis in Parkinson’s disease (as a consequence of l-dopa treatment) is best done with a very loose binding antipsychotic, such as clozapine or quetiapine, to allow for the low level of dopamine neurotransmission that is required for normal motor functioning to continue. Parkinson patients are dopamine-depleted, so it is very important not to block the little dopamine function that remains. The hypothesis is that atypical antipsychotic action (that is, low EPS and normal prolactin) occurs when endogenous dopamine is able to displace a loosely bound antipsychotic. This accords with the observation that low dosages of atypical antipsychotics are useful for Parkinson patients’.
It is well known in neurology that
l-dopa psychosis in a patient with Parkinson’s disease is best treated with a dosage of clozapine that is about 10% the dosage normally used for psychosis in schizophrenia. The “fast-off-D
2” hypothesis readily and quantitatively predicts this. As presented above, the antipsychotic dosage needed to occupy D
2 receptors is proportional to K ′ [1 + D / Dhigh], where K is the dissociation constant of the antipsychotic, D is the concentration of dopamine in the synaptic space during the momentary nerve impulse (equivalent to 200 nM, ref.
29), and where Dhigh is the dissociation constant of dopamine at the high-affinity state of D
2 (equivalent to 1.75 nM; Table 1). In Parkinson’s disease, where 90% to 95% of the dopamine content is absent, the value for D would be equivalent to 20 nM. Accordingly, the antipsychotic dosage for
l-dopa psychosis will be lower than that for schizophrenia psychosis by a factor of (1 + D / Dhigh) normal / (1 + D / Dhigh) Parkinson, or (1 + 200 / 1.75) / (1 + 20 / 1.75), or tenfold. Thus, while a daily dosage of 500 mg clozapine might be suitable for treating schizophrenia psychosis, a dosage of 50 mg (or less) would be more than adequate to treat
l-dopa psychosis. It is important to note that this calculation best holds for competition between endogenous dopamine and a loosely bound antipsychotic. A tightly bound antipsychotic such as haloperidol would not readily permit endogenous dopamine to replace it competitively.
Test of the “fast-off-D2” hypothesis using clozapine and isoclozapine
As reported by Kapur and others (
68), the single most powerful predictor of atypicality is the low affinity to, and fast dissociation from, the D
2 receptor—not high affinity to any other receptor. This hypothesis is supported by their findings that clozapine and isoclozapine have identical potencies on many cloned receptors (including muscarinic M1, dopamine D
1, dopamine D
4, 5-HT
1A, and 5-HT
2A receptors) but differ fivefold in their potency only on D
2 receptors. Thus, in several tests of atypicality (for example, early activation of certain genes, catalepsy in animals, and prolactin elevation), clozapine behaves like an atypical antipsychotic. Isoclozapine, however, behaves like a conventional antipsychotic.
Do antipsychotics elicit atypical action by blocking 5-HT receptors?
In addition to blocking dopamine receptors, the new atypical antipsychotic drugs also block 5-HT receptors. Although it has of ten been suggested that the blockade of 5-HT
2A receptors may alleviate the parkinsonism caused by D
2 blockade (
69,
70), most data do not support this principle.
Remoxipride is an important exception
Remoxipride is a highly effective atypical antipsychotic drug (not used in Canada) with no EPS and no hyperprolactinemia, yet it does not block 5-HT receptors.
5-HT blockade enhances catalepsy
Selective 5-HT
2A receptor blockade with the drug M100,907 markedly enhances, instead of reducing, the catalepsy (catalepsy in animals = EPS in humans) observed with submaximal dosages of the D
2 block by raclopride (
71).
No dopamine–5-HT correlation to cataleptic dosages
There is no correlation between the cataleptic dosages of neuroleptics and the ratio of the antipsychotic dissociation constants at D
2 and at 5-HT
2A receptors (
72,
73).
No sharp separation of typicals and atypicals
Using the ratio of antipsychotic dissociation constants obtained in our laboratory on human-cloned D2 and 5-HT2A receptors (Table 1), the demarcation between typical and atypical antipsychotics shown in Figure 4B is not sharp and is less clear than that found in Figure 4A. For example, of the 20 antipsychotics in Figure 4B, there are 3 to 4 apparent exceptions to the separation of typicals and atypicals. This compares to 2 or 3 apparent exceptions out of the 31 antipsychotics shown in Figure 4A. (Exceptions do not necessarily kill a theory, but explanations for the exceptions have to be found.)
No alleviation of extrapyramidal signs
A high degree of 5-HT
2A receptor occupancy (95%) by risperidone (6 mg daily) does not prevent EPS in 6 out of 7 patients (
74,
75).
5-HT block does not change D2 occupancies required
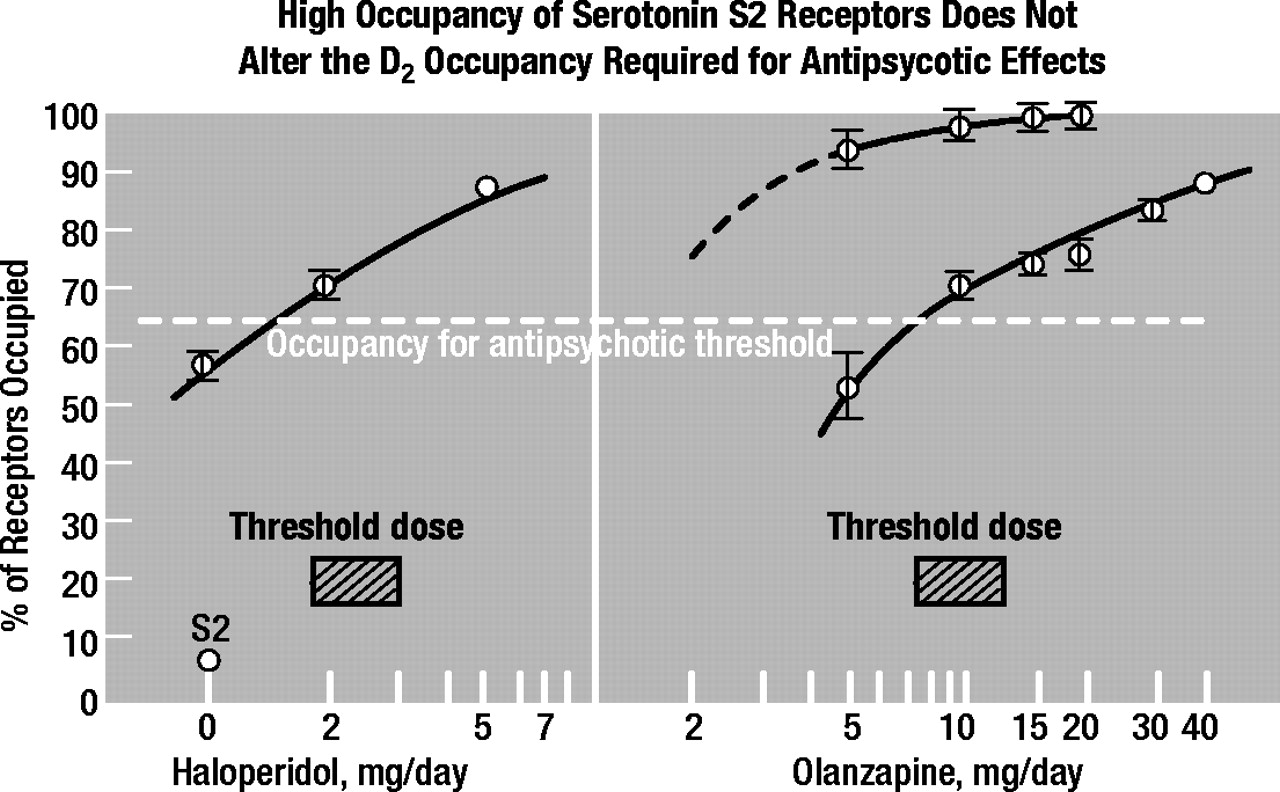
Using [
11C]raclopride for imaging brain D
2 receptors and [
11C]setoperone for imaging brain 5-HT
2A receptors, Kapur and others found that the high occupancy of 5-HT
2A receptors by olanzapine or by risperidone did not alter either the D
2 occupancy required for the antipsychotic effect or the D
2 occupancy at which EPS occur (
28). The threshold dosages for antipsychotic action consistently occupy 65% of brain D
2 receptors in patients, and the threshold dosages for EPS consistently occupy 80% of brain D
2 receptors in patients, whether or not the 5-HT
2A receptors are occupied. As illustrated in Figure 5, the results showed that the occupancy of D
2 receptors in first-episode schizophrenia patients was 65% for antipsychotic threshold dosages of haloperidol (1.5 to 2.1 mg daily) and olanzapine (7.5 to 10 mg daily), despite the negligible occupancy of 5-HT
2A receptors by haloperidol or the very high occupancy, exceeding 95%, by olanzapine. It is important to note that while first-episode patients may not be typical of patients with chronic schizophrenia, studies with the first-episode patients are exceedingly important in working out mechanisms of antipsychotic action, because they have had not previous exposure to drugs. It is not clear what clinical benefit, if any, is provided by the blockade of 5-HT receptors. Although low dosages of cyproheptadine have been used (
64) to block 5-HT
2A receptors and supplement antipsychotic administration, it should be noted that cyproheptadine has a D
2 blocking action. It has a K of 24 nM at D
2 receptors, compared with 63 nM for clozapine and 21 nM for amoxapine (Table 1). Amoxapine, though marketed as an antidepressant, has antipsychotic properties.
Amisulpride is an important exception
Amisulpride (used in Europe) is a highly effective antipsychotic that is atypical and does not occupy any 5-HT
2A receptors in humans at dosages up to 1200 mg daily (
76).
Chlorpromazine blocks 5-HT but elicits EPS
Chlorpromazine, the first typical antipsychotic, blocks 65% of 5-HT
2A receptors at 500 mg daily. This “high level of 5-HT
2A block . . . suggests that the distinct clinical profiles of chlorpromazine and clozapine are unrelated to 5-HT
2A receptor blockade” (
76).
5-HT block not needed for antipsychotic action
It has also been stated that the block of 5-HT
2A receptors “is not a prerequisite for the antipsychotic effect” (
74,
75). In fact, full block of 5-HT
2A receptors occurs at subtherapeutic dosages of risperidone, olanzapine, and clozapine, indicating that 5-HT
2A block has little or no antipsychotic action.
Do antipsychotics elicit atypical action by stimulating 5-HT receptors?
Although it has long been known that the stimulation of 5-HT
1A receptors in animals can alleviate catalepsy caused by D
2 blockade (
77,
78), there do not appear to be any antipsychotics that have this 5-HT
1A-stimulating action combined with D
2-blocking action.
It has recently been proposed that the stimulation of 5-HT
2A receptors by an inverse action is an important contribution to atypical antipsychotic action (
79). This is illustrated in Figure 4C, where the inverse stimulating potencies of antipsychotics on 5-HT
2A receptors are shown (from 79). However, because a few important atypical antipsychotics (including remoxipride and sulpiride) have no such stimulating action, it is unlikely that this feature contributes to atypical antipsychotic action. The data in Figure 4C do not reveal any clear demarcation between typicals and atypicals. Finally, Although the authors (
79) propose that M100,907 has the desired stimulating action, this compound has shown no antipsychotic activity in humans.
The future
Because brain imaging indicates that the traditional antipsychotics remain attached to dopamine D
2 receptors for at least 1 or 2 days, there is no rational need to medicate schizophrenia patients daily with typical antipsychotics. This reasoning has led to a new regimen of administering antipsychotics by “extended dosing,” wherein the patient receives a typical antipsychotic once every 3rd or 4th day (
80). Such a procedure, of course, could not be used with atypical antipsychotics because of their loose binding to D
2 and subsequent risk of relapse.
Clinicians can now apply this knowledge to the treatment of individual patients. Atypical agents, being newer and still protected by patent, are much more expensive than the older drugs. The fact that they do not elicit EPS and do not elevate prolactin levels does not mean that they are free of serious side effects. One could argue that the side effects associated with some of the atypical drugs (for example, agranulocytosis, obesity, diabetes, ophthalmological problems, cardiovascular problems, sexual problems, obsessive-compulsive symptoms, convulsions, and insomnia) are more serious than EPS, high prolactin, and even TD. Low-dose, extended-dosing regimens of typical drugs may be best suited for specific patients. Patients known to be nonadherent to regular medication may do better on those drugs that are more tightly bound to the D2 receptor, where risk of relapse through a short period of noncompliance is reduced. Conversely, patients with a history of neuroleptic malignant syndrome are best treated with drugs that are readily displaced, so that, should the syndrome return, the drug is quickly out of their brain. In patients with psychosis, high stress levels, accompanied by high endogenous dopamine release, will necessitate higher dosages of the antipsychotic drug. Periods of low stress will require lower dosages. Patients with psychosis who may temporarily benefit from high prolactin levels (for example, those who do not want to conceive or, conversely, postpartum women whose milk is in sufficient for breastfeeding) may preferentially be prescribed typical antipsychotics. On the other hand, typical antipsychotics should be discontinued in those with beginning signs of TD and the newer drugs prescribed instead. Knowing how drugs work greatly expands the clinician’s repertoire of strategies, allowing optimization of drug regimens for individualized treatment.