The last 20 years has seen the rise of “GeneTalk” (
1). A central phrase in GeneTalk, and one that has been heard widely in both lay (
2) and professional arenas, is “X is a gene for Y,” in which X is a particular gene on the human genome and Y is one of a wide variety of complex human disorders or traits such as depression, aggression, sexual orientation, obesity, infidelity, alcoholism, or schizophrenia.
This essay begins with a brief review of the historical origins of the concept of “a gene for”. I then propose criteria to assess the validity of this model of gene-phenotype relations and go on to evaluate these criteria as applied to genetic effects on psychiatric disorders. The essay concludes with general observations about our preconceptions and the reality of gene action in psychiatric disorders. Although many of the issues raised in this essay are equally applicable to etiologically complex medical disorders, the focus here will be on psychiatric illness.
Historical Origins of the Concept of “A Gene for”
Since humans started speculating about the nature of development and inheritance, a number of different conceptualizations have emerged about the nature of the guiding forces in these processes (
3). In the 20th century, this discourse has come to focus largely on the nature of what Mendel originally termed “anlagen” or “elements,” which in 1909 became “genes” (
4).
Of the multiple different views of the nature of the “gene,” the one in which we are interested—a gene defined by the phenotype that it causes—originated in the developmental theory of preformationism (
5). One of the earliest articulated theories of development, preformationism was first proposed by Aristotle but became particularly influential in the 17th century (
3,
5,
6). The essentials of the theory are eloquently described by Jacob:
At a time when living beings are known by their visible structure alone, what has to be explained about generation [i.e., development] is the maintenance of this primary structure through succeeding generations. The structure cannot itself disappear; it has to persist in the seed from one generation to another. To maintain the continuity of shape, the “germ” of the little being to come has to be contained in the seed; it has to be “preformed.” The germ already represents the visible structure of the future child. . .It is the plan for the future living body. . .already materialized, like a miniature of the organism to come. It is like a scale model with all the parts, pieces and details already in position. . .Fertilization only activates it and starts it growing. Only then can the germ develop, expand in all directions and acquire its final size, like those Japanese paper flowers which, when placed in water, unwind, unfold and assume their final shape. (7, p. 57)
In preformationism, the egg or sperm was understood to contain all the final traits of the mature organism. Development consisted of the expansion of these preformed characteristics (or anlagen) into the individual traits of the adult organism. That is, these anlagen were truly for the adult traits with which they had a simple and direct causal relationship.
In the 19th century, as the young field of biology struggled to fathom the mechanism of transmission of traits across generations, a number of the proposed theories of inheritance (where the “units” of inheritance had names such as pangenes, stirps, and gemmules) had important preformationist themes (
3,
4). When Mendel’s groundbreaking work on genetics (originally published in 1866) was rediscovered in 1900, one common interpretation was that his “elements of inheritance” were the discrete anlagen predicted by preformationist theories (
5). This interpretation was favored by two of the most influential geneticists of the day, the Dutchman de Vries (the most famous of the three “co-rediscovers” of Mendel [4]) and the Englishman Bateson (
8).
In summarizing this exciting period in the history of biology, Allen (
9) writes
The implications that the discreteness of the gene implied the organism was constructed as a “mosaic” of adult traits was given explicit voice by Bateson with the first years of his encounter with Mendelism.
Allen goes on to quote two passages from Bateson written, respectively, in 1901 and 1902 (
9):
In so far as Mendel’s law applies, the conclusion is forced upon us that the living organism is a complex of characteristics of which some, at least, are dissociable and are capable of being replicated by others. We thus reach the conception of unit characters which may be rearranged in the formation of reproductive cells.
The organism is a collection of traits. We can pull out the yellowness and plug in greenness, pull out tallness and plug in dwarfness.
Bateson was recasting, in a new language, preformationist concepts. The Mendelian anlagen (later genes) could be defined by their relationship to the particular phenotype (or “unit character”) with which it had a privileged causal link. That is, such genes caused phenotypes in the same way that the preformationist anlagen prefigured adult traits. From this perspective, it made sense to speak of “a gene for greenness,” “a gene for tallness,” or a gene for any of the other innumerable unit characteristics of the adult organism. It is in this context that a rarely discussed early chapter of psychiatric genetics in the United States must be viewed, when reports appeared claiming to find, in series of large pedigrees, evidence for Mendelian genes “for” “Nomadism or the wandering impulse” (
10) and “the neuropathic constitution” (
11).
This preformationist concept of the gene proved attractive to medical geneticists who, over the course of the 20th century, showed that most classical genetic disorders in humans (termed “Mendelian” diseases in honor of the Austrian monk) were due to hereditary units that behaved just like those first examined by Mendel (
12).
While medical geneticists came to understand that in biological systems, genes actually code for proteins, it became convenient and seemingly natural to think about preformationist-like genes for these classical genetic diseases in humans.
The last 30 years have seen three interrelated further themes in the “a gene for” story. First, in the mid-1970s two influential books appeared that heightened the profile of genes and their potential impact on human behavior. “Sociobiology: The New Synthesis” by Wilson (
13) launched the field of sociobiology (and later evolutionary psychology), discourse in which commonly included the concept of “genes for” a wide range of traits, including altruism, territoriality, jealousy, and ethics. “The Selfish Gene” by Dawkins (
14) proposed a gene-centered view of evolution in which an organism, with its wide array of phenotypes, was viewed as a vehicle through which genes replicate themselves over evolutionary time. Second, with the developmental of an ever increasing set of powerful molecular tools, the specific genes and then the specific mutations in those genes were discovered that were responsible for all major classic human genetic disorders. So, when speaking about “a gene for Y” in which Y was sickle-cell anemia, cystic fibrosis, or Huntington’s chorea it became possible to conceive of the gene not only as an abstract transmitted “unit” but also as a discrete piece of DNA at a specific location on a chromosome. Third, prompted by the sequencing of the human genome, the concept that DNA represented the “blueprint” of life (or in related versions the “code” or “recipe” for life) was widely promulgated in both the scientific and lay literature (
2). The preformationist themes in this metaphor are evident: genes are to phenotypes as blueprints of a building are to the building themselves.
So, this historical sketch suggests that our current concept of “X is a gene for Y” in humans has four major interrelated historical roots. First, the concept that development anlagen could be “for” adult traits arose in preformationist developmental theories. Second, the discovery of Mendel’s “elements” was interpreted by some as verifying this concept. Third, the idea that genes could be “for” human traits was supported by the discovery that genes for classical Mendelian medical disorders often acted just like the hereditary elements found in Mendel’s pea plants. Finally, these concepts became linked to DNA by a series of stunning discoveries in the last 20 years, so that strength of the “icon” of the double helix provided particular luster to potential discoveries in psychiatry of “a gene for”.
Criteria for the Concept of “A Gene for”
The remainder of this essay addresses the question of whether this preformationist model of gene action—in which genes are “for” phenotypes—is appropriate for psychiatry. Based in part on prior efforts to develop guidelines for causal inference in epidemiology (e.g., reference
15), I suggest five criteria by which to judge the validity of the claim “X is a gene for Y”: 1) strength of association of X with Y, 2) specificity of relationship of X with Y, 3) non-contingency of the effect of X on Y, 4) causal proximity of X to Y, and 5) the degree to which X is the appropriate level of explanation for Y. In sum, I argue that
If gene X has a strong, specific association with disease Y in all known environments and the physiological pathway from X to Y is short or well-understood, then it may be appropriate to speak of X as a gene for Y.
But first, a few details are needed. The scientific basis of most claims that “X is a gene for Y” results from a statistical test called association analysis. In its simplest form, this test compares the frequency of specific DNA variants in or around gene X in a set of cases with disorder Y and a set of matched control subjects. An association is claimed if the frequency of these variants differ significantly in cases and control subjects. In both a conceptual and statistical sense, this approach is no different from the methods commonly used in the biomedical and social sciences to assess the relationship between putative risk factors and outcome variables such as smoking and lung cancer or childhood sexual abuse and depression.
Therefore, standard “a gene for” claims are based on statistical and not biological grounds. Biological studies that trace etiologic pathways from X to Y should follow claims for association and would certainly provide confirmatory data. However, they have been very rare to date in psychiatric genetics. On its own, a significant p value in an association study tells you nothing about the nature of the causal relationship between the gene and the disease.
Strength of Association
As with any risk factor for any outcome, the strength of association between a specific gene and a particular disease can vary in magnitude. In considering the criteria for “a gene for. . .,” an historical standard of comparison is what has come to be called a Mendelian gene. The action of Mendelian genes is deterministic and not probabilistic. If a plant inherits a particular copy of the gene for wrinkled peas, it would not matter how much sunshine the plant received or the quality of its fertilizer. The plant will have wrinkled peas no matter what the environment does. In humans, we have many diseases that are due to Mendelian genes that behave exactly like the genes Mendel studied in his pea plants (
12). If you have one copy of the pathogenic gene for Huntington’s disease, it does not matter what your diet is, whether your parents were loving or harsh, or if your peer group in adolescence were boy scouts or petty criminals. If you have the mutated gene and you live long enough, you will develop the disease.
Furthermore, for most Mendelian genes in man, the only way to get the disorder is to have the disease gene. There is no way to “acquire” cystic fibrosis or Huntington’s disease through environmental exposure. So if having the disease gene always produces the disorder and the disorder never occurs without the disease gene, this produces a perfect association between the disease gene (X) and the disorder (Y). (Reality is somewhat more complex. Most Mendelian genes in man contain several different mutations, each of which can cause diseases that are sometimes of quite variable severity. But this claim still holds for all mutations of the gene considered together.)
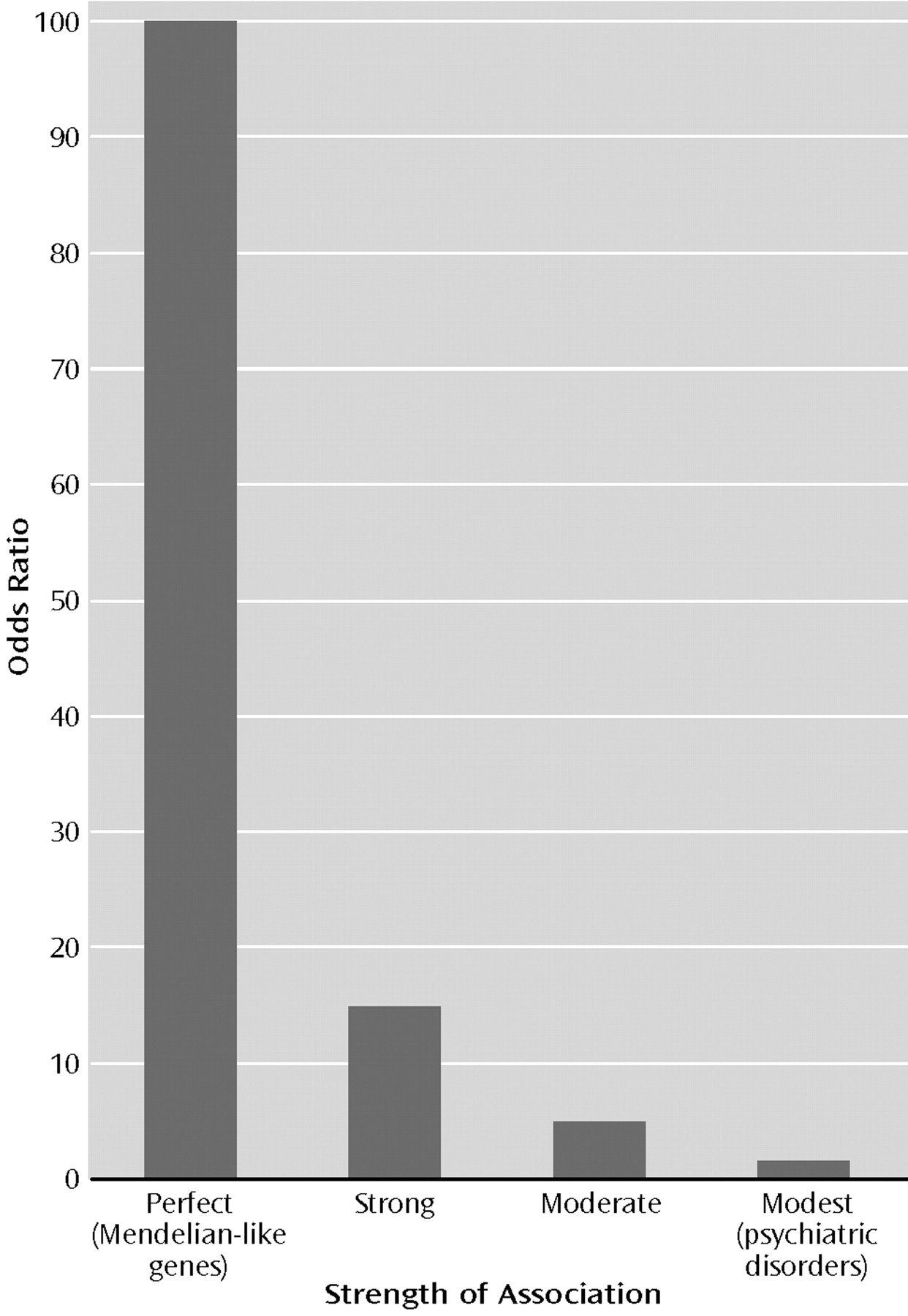
The strength of an association between a risk factor and a disease is most frequently quantified by a statistic called the odds ratio. Formally, the odds ratio is defined as the ratio of the odds of developing the disease among those exposed to the risk factor and the odds of disease among those not exposed to the risk factor. For Mendelian disorders in man, since the first of these figures is one and the second is zero, the odds ratio for the disorder given the pathogenic gene is infinite. Since this is a rather stringent criteria, for the sake of argument, let us say the association with Mendelian-like genes (an historical model for the concept of “a gene for”) has an odds ratio of approximately 100 (Figure 1).
Are there any genes whose strength of association with a psychiatric disorder is Mendelian-like? Two related sources of information, both gathered in the last two decades, indicate that the answer to this question is almost certainly “No.” First, a gene that has a deterministic or nearly deterministic relationship with a phenotype produces an unmistakable signature in the pattern of illness in large pedigrees. Numerous investigators have now searched many parts of the globe (including nearly all psychiatric facilities in a modest-sized country [16]) seeking pedigrees in which major forms of psychiatric illness—especially schizophrenia and bipolar illness—are distributed in the pattern expected from a Mendelian-like gene. Such pedigrees have not been found.
Second, Mendelian-like genes also produce a distinctive result in genome-wide linkage studies, which effectively sweep the human genome looking for regions that contain genes that have an impact on risk of illness. While the technical details need not concern us, experts agree that for those disorders studied in genome-wide linkage scans of reasonable size and quality—especially schizophrenia, bipolar illness, panic disorder, and eating disorders—conclusive evidence has accumulated that even moderately rare genes of Mendelian-like effect do not exist. (The available evidence does not permit us to rule out, however, very rare Mendelian-like genes.)
So, if we lack Mendelian genes for psychiatric disorders, with their very high odds ratios, what sort of magnitude of associations might we expect? One set of benchmarks might be provided by three examples of what would be considered very strong associations in epidemiology. The estimated odds ratio between heavy smoking and small cell carcinoma of the lung is approximately 20 (
17), between industrial exposure to asbestos and mesothelioma is approximately 15 (
18), and between severe stressful life events and the onset of major depression is approximately 12 (
19).
Another more modest benchmark is provided by the two outstanding genetic association results in neuropsychiatry of the last decades. The association between the pathogenic “4 allele” of the apolipoprotein E gene and Alzheimer’s disease produces, in Caucasian populations, an odds ratio of approximately 3.0 (
20). In Asian populations, the possession of the slow-metabolizing (ALDH2*2) copy of the aldehyde dehydrogenase gene conveys up to a 10-fold reduction in risk for the development of alcoholism (
21).
So, as depicted in Figure 1, we have three possible benchmarks for the strength of the gene-phenotype association for psychiatric disorders: Mendelian-like (odds ratio of approximately 100), strong (odds ratio=12–20), or moderate (odds ratio=3–10).
Trying to summarize the magnitude of association found between functional candidate genes and psychiatric disorders is problematic because of the multiple methodologic difficulties in the interpretation of such studies (
22–
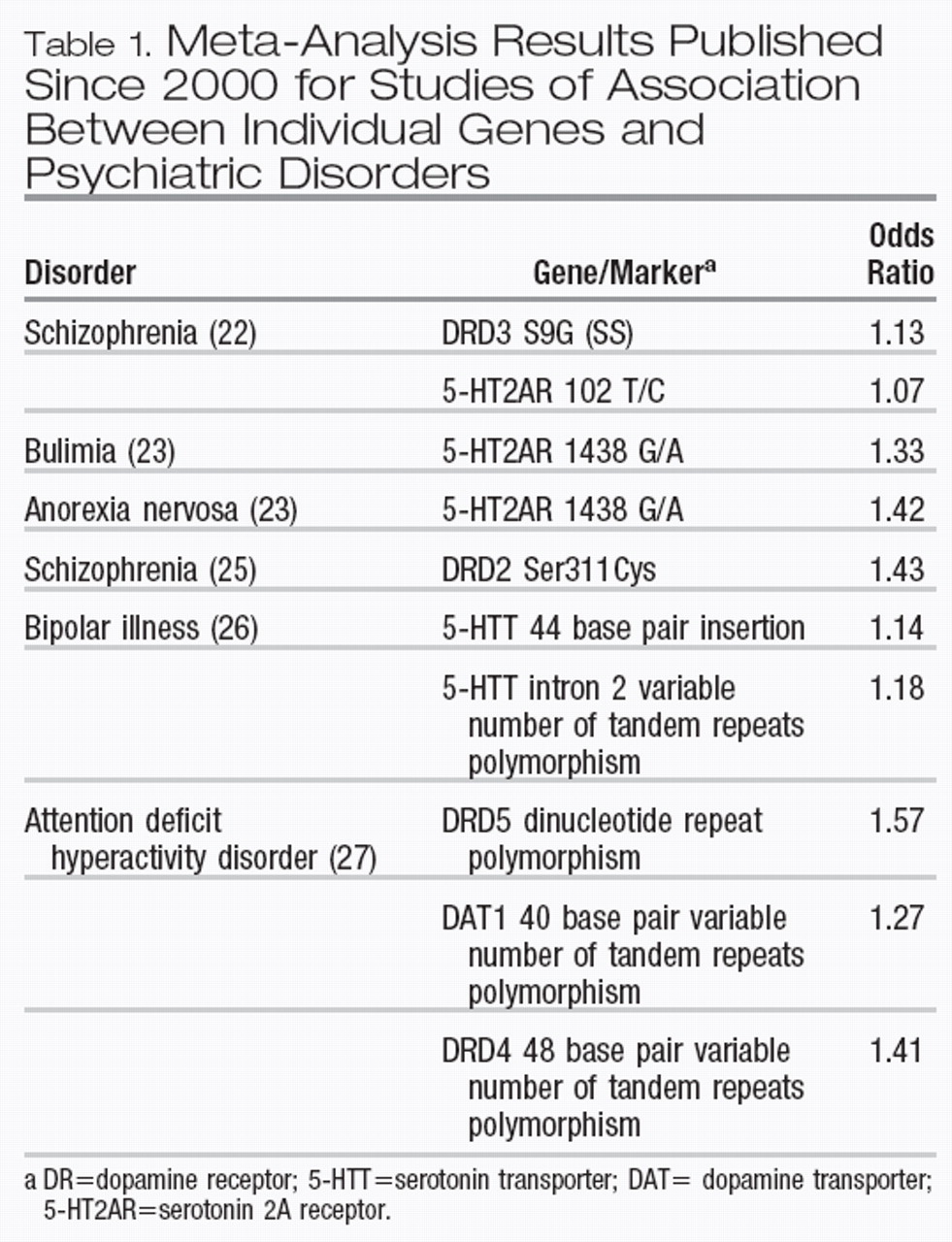
24). Greatest reliability should be placed on the results of meta-analyses, which are now beginning to appear in the literature. A PubMed search from 2000 on (using publication type of “metaanalysis” and search words “gene” and “association”) followed by a hand search and elimination of duplication yielded 10 significant meta-analytic estimates of odds ratios between individual genes and psychiatric disorders (Table 1) (excluding results from those meta-analyses that did not support the original positive reports). The odds ratios ranged from 1.07 to 1.57 with a median of approximately 1.30.
Another strategy to localize candidate genes is to look for them under linkage peaks (so-called positional candidate genes). In schizophrenia, replicated evidence is now emerging for several such genes (
28). For these genes, disease-associated haplotypes—small sections of DNA that have traveled together over evolutionary time—can often be found. The two best replicated positional candidate genes for schizophrenia are dysbindin 1 and neuregulin 1. Not counting the original reports (where the effect size might be biased upward), estimates are available for the association between high-risk haplotypes and schizophrenia for both of these genes. For dysbindin, odds ratios of 1.24 (
29), 1.23 (
30), 1.40 (
31), 1.70 (
32), and 1.58 (
33) have been reported or calculated from replication reports. For neuregulin 1, two replications were noted in a recent review, with odds ratios estimated to be 1.25 and 1.80 (
28).
Taken together, the meta-analyses of functional candidate gene association studies and early results from positional candidate genes suggest that the magnitude of the associations between individual genes and psychiatric illnesses have small odds ratios, largely from 1.1 to 1.6. Compared to our benchmarks, this effect size is very modest (Figure 1). Perhaps genes (or particular mutations or haplotypes) of larger effect size will be found. While results from linkage studies suggest that this is unlikely, it cannot be ruled out. Also to be considered is the statistical dictum that the first set of effects detected in any research area tend to be the most robust. If this is correct, further genes discovered for psychiatric disorders are likely to have smaller average effects than the genes found to date. The preformationist concept of “a gene for” implied a predetermined and largely irrevocable link between gene and phenotype. This is the pattern of association observed between gene and phenotype from Mendel’s original traits and for Mendelian genetic disorders in humans. By contrast, for psychiatric disorders, individual genes appear to have a quite modest association with psychiatric illness. While they may have an impact on risk, individual genes hardly predetermine illness, as would be expected if we had discovered “genes for” mental disorders.
Specificity of Association
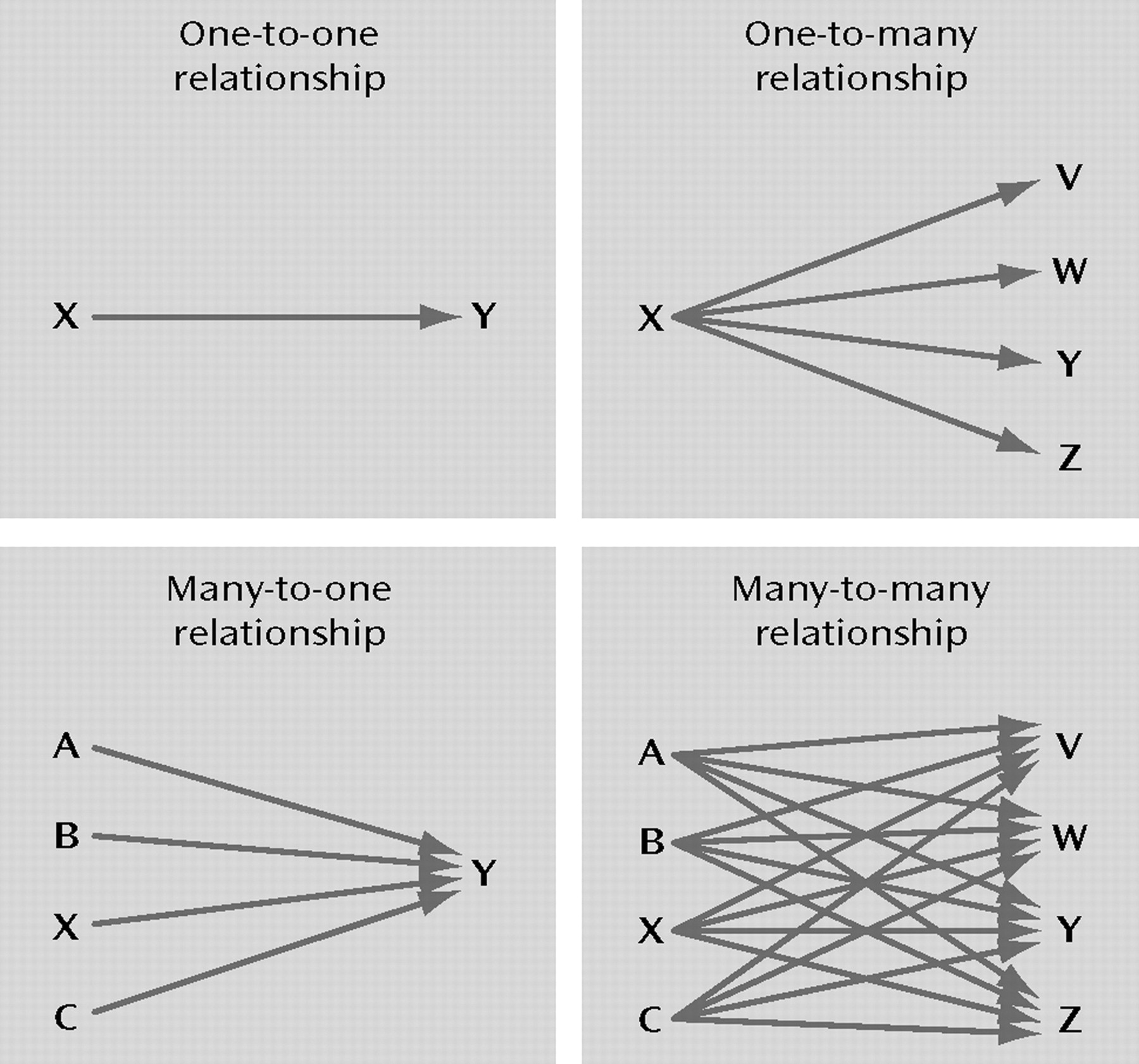
The second criterion to evaluate the appropriateness of the concept of “X is a gene for Y” is the degree of specificity in the relationship between X and Y. As illustrated in Figure 2, does X influence risk for any other disorders in addition to Y? Or are there other genes that contribute to Y in addition to X?
In preformationist theory, anlagen had highly specific associations with the adult traits into which they developed. The hereditary elements of the pea that Mendel studied also had quite specific phenotypic effects. That is, one gene influenced pea color but not shape or height while another influenced shape but not height or color. However, as genetics developed, many genes were found that impacted on a variety of phenotypic characteristics—a phenomenon called pleiotropy.
In man, many Mendelian genes produce one and only one disease syndrome (although sometimes of varying severity depending on the specific mutation). But there are exceptions where different abnormalities in a single gene can produce distinct genetic diseases.
How specific are individual genes in their impact on risk for psychiatric disorders? Do most genes influence risk for one and only one psychiatric disorder? Twin studies, which study “genes” in the aggregate, suggest that genetic risk factors for psychiatric disorders are often nonspecific in their effect. A large-scale twin study of seven psychiatric and substance use disorders found one common genetic risk factor predisposing to drug abuse, alcohol dependence, antisocial personality disorder, and conduct disorder and a second common genetic factor influencing risk for major depression, generalized anxiety disorder, and phobia (
34). Overlap of genetic risk factors for multiple disorders have been demonstrated in other twin studies (e.g., references
35–
37).
We know much less about the specificity of the spectrum of effects on psychiatric disorders of individual genes. Meta-analyses reviewed in Table 1 show that variants at one gene (the 5-HT2A receptor) may predispose to risk for three different disorders (schizophrenia, bulimia, and anorexia nervosa). A pair of overlapping genes on chromosome 13q (termed G30 and G72) may be associated both with schizophrenia and bipolar illness (
28). A number of overlapping positive regions in linkage genome scans for bipolar illness and schizophrenia have led some to argue that this reflects shared genes between these two disorders (
38). While difficult to evaluate critically, claims have been made that several popular candidate genes (e.g., serotonin transporter, dopamine transporter, dopamine 2 receptor) are significantly associated with a wide variety of psychiatric disorders or psychiatrically relevant traits (
39,
40). While much remains unknown, current evidence suggests that many genes that influence risk for psychiatric disorders will not be diagnostically specific in their effect, thereby resembling the one-to-many relationship in Figure 2 rather than the one-to-one relationship.
We are on firmer ground in evaluating whether genetic risk for psychiatric disorders results from the action of a single gene (the one-to-one relationship in Figure 2) or multiple genes (the many-to-one relationship in Figure 2). Some evidence bears on this question indirectly, as follows. Twin and adoption studies provide convincing evidence for significant genetic effects on virtually all major psychiatric disorders (
41). Therefore, genes that affect risk for these disorders must exist somewhere on the human genome. Linkage studies examine how these aggregate genetic risk factors are distributed across the genome. If genetic risk resulted from a single gene, then all the linkage “signal” would be concentrated in a single location, with a resulting clear and robust statistical linkage peak. But, as noted earlier, this is a pattern that has not been observed in published genome scans for psychiatric disorders. Instead, a number of modest linkage peaks are usually seen, suggesting that the “packets” of genetic risk for these disorders are widely dispersed across the genome. (To complicate matters, genome scans will underestimate the number of genomic regions involved because of low power to detect genes of small effect size, but will overestimate the number because some of the observed “peaks” will be false positives.)
Recently, data have emerged that addresses this question directly. A careful meta-analysis of 20 genome scans for schizophrenia has suggested 10 genomic regions likely to contain susceptibility genes (
42). In addition, current evidence of bipolar disorder, the second-best-studied psychiatric disorder by linkage scans, also suggests multiple loci (
43).
The specificity of association implied in the “a gene for” concept has another implication worth exploring. Consistent with preformationist theory, specificity of gene action implies that the gene contains all information needed for the development of the trait. The environment might impact on the final phenotype, but its effect is nonspecific. That is, the gene “codes for” the trait, while the environment reflects background factors that support development but is not in and of itself “information-carrying.”
To illustrate how commonly we see genes and environment in this light, it is worth pondering a curious and asymmetrical feature of GeneTalk. While we find it easy to use the phrase “X is a gene for Y,” it feels quite odd to say “A is an environment for B.” For example, a large body of empirical work supports the hypothesis that severe life events are important environmental risk factors for major depression (
44). The magnitude of the association between such events and the subsequent depressive episode is far greater than that observed for any of the genes that we have reviewed here. Yet, who has heard the phrase “a romantic breakup is an environment for depression”? I suggest that we feel comfortable with “X is a gene for Y” and not “A is an environment for B” because we implicitly assume that genes have a privileged causal relationship with the phenotype not shared by environmental factors.
However, empirical evidence does not support the position that genes code specifically for psychiatric illness while the environment reflects nonspecific “background effects.” By definition, environmental factors are central to the etiology of posttraumatic stress disorder. In the aforementioned multivariate twin model, what distinguished major depression, generalized anxiety disorder, and phobia from one another were environmental and not genetic risk factors (
34). In a detailed study of the impact of childhood parental loss on risk for common psychiatric and substance use disorders, death of a parent was specific in increasing risk for major depression and no other disorder (Kendler et al., unpublished results). Consistent with studies of stressful life events that have shown moderate separation of depressogenic and anxiogenic events (
45,
46), a multivariate genetic study of symptoms of anxiety and depression showed that genetic factors influence nonspecific risk for all symptoms, whereas two environmental factors were identified that predisposed, with moderate specificity, for symptoms of depression and anxiety, respectively (
47).
The preformationist concept of “a gene for” implies high levels of specificity between gene and phenotype. While much remains to be learned in this area, current evidence suggests that instead of the “one-to-one” relationship implied by the concept of “a gene for. . .,” genes and disorders in psychiatry are likely to have the “many-to-many” relationship depicted in Figure 2.
(The evidence that the association between individual genes and psychiatric disorders are typically weak and may often be nonspecific does not mean that the identification of such genes is unimportant. For example, such discoveries can identify pathophysiologic pathways, begin the lengthy process of clarifying how individual genes interact with each other and with environmental exposures to produce illness, and provide new targets for treatment.)
Noncontingency of Association
Noncontingent association means that the relationship between gene X and disorder Y is not dependent on other factors, particularly exposure to a specific environment or on the presence of other genes. As mentioned earlier, this is a typical (albeit not uniform) feature of genes that cause classical Mendelian disorders in humans. If the association between gene and disease were contingent on particular environmental exposures, then we would have to amend our statement to read “X is a gene for Y given exposure to environment Z.”
Environmental contingencies for genetic effects on psychiatric disorders have been little investigated. Twin and adoption studies suggest that the impact of aggregate “genes” for major depression are altered by exposure to stressful life events (
19,
48) and for schizophrenia and conduct disorder by exposure to a dysfunctional rearing environment (
49,
50). A range of twin studies suggest that environmental experiences have an impact on genetic risk for several psychiatrically relevant traits, including aggression, disinhibition, and smoking (
51). Recently, Caspi and colleagues have found evidence for interactions between environmental risk factors and particular genes in the production of antisocial behavior (
52) and depression (
53), with the former finding having been replicated (
54).
We know almost nothing about gene-by-gene interactions in the etiology of psychiatric disorders. Although a number of association studies have reported interactions, I am unaware of any that have been widely replicated or supported by meta-analyses. Using statistical models applied to risk of illness in various classes of relatives, Risch has claimed that gene-by-gene interactions are important in the etiology of schizophrenia (
55).
Overall, we know little about the contingent nature of genetic effects for psychiatric disorders. The available information suggests that gene action contingent upon certain environmental exposures is probably not rare and may be relatively common for psychiatric disorders. This is also inconsistent with the preformationist concept of “a gene for. . .”.
Causal Proximity
Preformationist developmental models assumed that anlagen developed directly into adult traits. The “blueprint for life” metaphor similarly assumes a direct correspondence between individual parts of the blueprint (windows, doors, fixtures) and the corresponding units of the completed building. Conceptualizing genes in this preformationist framework therefore carries the implicit assumption of a direct causal link between gene and phenotype. It is only with this assumption that usage of the “a gene for. . .” is congruent with the common meaning of the phrase “X is for Y” in English. To clarify this point, let’s examine a typical list of such statements:
I use a knife for buttering my toast.
I have a backpack for carrying my computer to work each day.
I was upset at my son for not doing his chores.
In each case, there is an implied direct and immediate relationship between X and Y. To put it more formally, X and Y are directly linked in a formal logical train of action (first two examples) or thought (third example).
Now, how does this common sense meaning of the word “for” apply to the phrase “X is a gene for Y”? Let me illustrate the problem with a vignette
A jumbo jet contains about as many parts as there are genes in the human genome. If someone went into the fuselage and removed a 2-foot length of hydraulic cable connecting the cockpit to the wing flaps, the plane could not take off. Is this piece of equipment then a cable for flying?
Most of us would be uneasy answering yes to this question. Why? Because this example violates our conception of causal proximity. When we say X is for Y, we expect X to be, to a first approximation, directly and immediately related to Y. That is not the case for the cable and flying. There are many, many mechanical steps required to get from the function of that cable to a jumbo jet rising off the runway.
Another vignette:
Assume a Mendelian genetic disease due to a mutation in gene K. Gene K’s normal function is to produce an enzyme L that breaks down metabolite M in cells allowing M to be harmlessly secreted from the body. When K has a pathogenic mutation, the enzyme L that is produced no longer works. Therefore, levels of M rise, producing a well understood series of toxic effects, thereby producing the genetic disorder N.
This scenario suggests the following potentially simple causal chain: mutated gene K→dysfunctional enzyme L→excess metabolite M→disorder N. In this admittedly oversimplified story, a case could be made that gene K had sufficient causal proximity to disorder N to make plausible the claim that “K is a gene for N.” However, it might be argued that even here, the complexity of the paths from levels of M to disorder N may be far from “simple.”
Contrast this situation to the causal chain from a gene mutation to a complex psychiatric disorder such as schizophrenia. Although early efforts have been made to begin to trace such pathways (e.g., reference
56), we probably do not know enough to articulate all the specific causal steps that would be needed to go from DNA basepair variation to, for example, the cognitive processes that predispose to delusion formation. What we can conclude with some confidence is that it will be very complex. Indeed, the causal link between that hydraulic cable and the jumbo jet flying will probably look very simple and short compared to the causal relationship between individual genes and the manifestations of schizophrenia. While the nature of the evidence reviewed here is largely inferential, it suggests that the pathways from most genes for psychiatric illness to their phenotypes would fail the causal proximity criterion implicit in the concept of “X is a gene for Y.”
Appropriate Level of Explanation
Scientific theories typically strive to explain phenomenon at the most informative level. To provide an absurd example, no one would seek to understand the origin of hypertension at the level of quarks. In some ultimate way, quarks may be involved. But quarks are just the wrong level of inquiry for the problem.
To illustrate how this issue—the appropriateness of level of explanation—may apply to our evaluation of the concept of “a gene for. . .” consider these two “thought experiments”:
Defects in gene X produce such profound mental retardation that affected individuals never develop speech. Is X is a gene for language?
A research group has localized a gene that controls development of perfect pitch (57). Assuming that individuals with perfect pitch tend to particularly appreciate the music of Mozart, should they declare that they have found a gene for liking Mozart? For the first scenario, the answer to the query is clearly “No.” Although gene X is associated with an absence of language development, its phenotypic effects are best understood at the level of mental retardation, with muteness as a nonspecific consequence. X might be a “gene for” mental retardation but not language.
Although the second scenario is subtler, if the causal pathway is truly gene variant→pitch perception→liking Mozart, then it is better science to conclude that this is a gene that influences pitch perception, one of the many effects of which might be to alter the pleasure of listening to Mozart. It is better science because it is more parsimonious (this gene is likely to have other effects such as influencing the pleasure of listening to Haydn, Beethoven, and Brahms) and because it has greater explanatory power.
A final scenario:
Scientist A studied the behavioral correlates of a particular variant at gene X and concluded “This is a very interesting gene that increases the rates of sky diving, speeding, mountain climbing, bungee jumping, and unprotected casual sex.” Scientist B studied the same variant and concluded “This is a very interesting gene and effects levels of sensation-seeking.”
Who has done the better science? Since sensation seeking (and its close cousin novelty-seeking) are well studied traits (
41), scientist B has provided results that are more parsimonious and potentially provide greater explanatory power. For example, only scientist B could predict that this gene ought to be related to other behaviors, like drug taking, that are known to be correlated with sensation-seeking.
As reviewed here, genes have been and will continue to be found that have statistical relationships with risk for psychiatric disorders. However, will the action of these genes be best explained at the level of the disorders themselves? While we cannot answer this question definitively, I would judge this to be unlikely. Far more plausible is that we will find genes whose mode of action can be best understood at the level of more basic biological processes (e.g., neuronal cell migrations during development) and/or mental functions (e.g., processing of threat stimuli).
Overview and Conclusion
The goal of this essay is to understand the historical origins of the key phrase “X is a gene for Y” and then to evaluate its appropriateness for psychiatric disorders. Our interest, of course, is not merely the phrase itself, but the conceptual framework that underlies this form of Gene-Talk. The use of the phrase “a gene for” implies (and in fact only makes sense in the context of) genes which—like preformationist anlagen—“code for” psychiatric illness in a simple, direct, and powerful way.
I argue that the concept of “a gene for. . .” can best be understood as deriving from preformationist developmental theory which, in turn, influenced the interpretation of the concept of a gene in the work of Mendel, in medical genetics, and most recently in human molecular genetics. Five criteria were proposed for evaluating whether the preformationist concept of “X is a gene for Y” is appropriate for psychiatric disorders. I then reviewed the available evidence, which was of variable quality, that addressed each of these criteria.
The strength of association between individual genes and psychiatric disorders is weak and often nonspecific. Genes do not appear to contain all the information needed for the development of psychiatric illness, since environmental factors have, for several disorders, been shown to have causal specificity. The action of genes on psychiatric disorders may frequently be contingent on environmental exposures, although much needs to be learned in this area. The causal chain from genes to psychiatric disorders is probably long and complex. The appropriate level of explanation for gene action is much more likely to be basic biological or mental processes that contribute to psychiatric disorders rather than the disorders themselves. Thus, with varying degrees of confidence, the genetic contribution to psychiatric disorders fails to meet any of the five criteria for the preformationist concept of “a gene for. . .”. The impact of individual genes on risk for psychiatric illness is small, often nonspecific, and embedded in causal pathways of stunning complexity.
On this basis, I suggest that we conclude that the phrase “X is a gene for Y,” and the preformationist concept of gene action that underlies it, are inappropriate for psychiatric disorders. The strong, clear, and direct causal relationship implied by the concept of “a gene for. . .” does not exist for psychiatric disorders. Although we may wish it to be true, we do not have and are not likely to ever discover “genes for” psychiatric illness.